Biosimilars: Ein etabliertes Konzept und praktische Erfahrungen aus der Anwendung
Interessengebiete: Allgemeinmedizin und Innere Medizin, Hals-Nasen-Ohrenheilkunde, Haut- und Geschlechtskrankheiten, Neurologie, Gastroenterologie, Immunologie, Rheumatologie
Seit der Erstzulassung vor rund 40 Jahren haben Biologika die Therapie in vielen Indikationen revolutioniert und stellen heute unverzichtbare Standardtherapeutika dar. Die ersten Biosimilars kamen als Nachfolgeprodukte von Biologika 2006 auf den Markt. Die Entwicklung von Biosimilars ist generell deutlich aufwändiger gegenüber Generika und unterliegt vielen Regularien. Die zunehmende Anzahl und Akzeptanz von Biosimilars hat vor allem in den letzten Jahren zu erheblichen Kosteneinsparungen im Gesundheitssystem geführt und die Versorgung der Patienten deutlich verbessert. In den nächsten Jahren laufen zahlreiche Patente von Biologika aus, so dass sich dieser Trend auch in Zukunft fortsetzen wird.
In dieser Fortbildung erfahren Sie u. a., was Mikroheterogenität bei biologischen Arzneimitteln bedeutet, unter welchen Voraussetzungen die Extrapolation von klinischen Daten erlaubt ist, welche klinischen Erfahrungen mit Biosimilars in den letzten 15 Jahren gesammelt wurden und wie die europäische Zulassungsbehörde, EMA, die aktuelle Sicherheitslage zur Austauschbarkeit von Biosimilars bewertet. Außerdem erhalten Sie praktische Tipps für eine positive Arzt-Patienten-Kommunikation, die Nocebo-Effekte bei Umstellung auf Biosimilars minimieren und somit den Therapieerfolg erhöhen kann.
Wenn Sie das Video zur arztCME-Fortbildung herunterladen wollen, registrieren Sie sich bitte oder melden Sie sich an.
Kursinhalt
Inhaltsverzeichnis
Kursbeschreibung
Seit der Erstzulassung vor rund 40 Jahren haben Biologika die Therapie in vielen Indikationen revolutioniert und stellen heute unverzichtbare Standardtherapeutika dar. Die ersten Biosimilars kamen als Nachfolgeprodukte von Biologika 2006 auf den Markt. Die Entwicklung von Biosimilars ist generell deutlich aufwändiger gegenüber Generika und unterliegt vielen Regularien. Die zunehmende Anzahl und Akzeptanz von Biosimilars hat vor allem in den letzten Jahren zu erheblichen Kosteneinsparungen im Gesundheitssystem geführt. In den nächsten Jahren laufen zahlreiche Patente von Biologika aus, so dass sich dieser Trend auch in Zukunft fortsetzen wird.
In dieser Fortbildung erfahren Sie u. a., was Mikroheterogenität bei biologischen Arzneimitteln bedeutet, unter welchen Voraussetzungen die Extrapolation von klinischen Daten erlaubt ist, welche klinischen Erfahrungen mit Biosimilars in den letzten 15 Jahren gesammelt wurden und wie die europäische Zulassungsbehörde, EMA, die aktuelle Sicherheitslage zur Austauschbarkeit von Biosimilars bewertet. Außerdem erhalten Sie praktische Tipps für eine positive Arzt-Patienten-Kommunikation, die Nocebo-Effekte bei Umstellung auf Biosimilars minimieren und somit den Therapieerfolg erhöhen kann.
Biologika: Historie und klinische Bedeutung
Seit etwa 40 Jahren gibt es klinische Erfahrungen mit Biologika in der Arzneimitteltherapie. In Deutschland wurden im Jahr 1982 die ersten rekombinanten Humaninsulin-Prä- parate zur Behandlung von Diabetes zugelassen [1]. Darauf folgten weitere Hormone und Wachstumsfaktoren, wie z. B. menschliches Wachstumshormon gegen Minderwuchs [1]. Ende der 1990er Jahre wurden die ersten monoklonalen Antikörper zur Behandlung von Krebs- und Autoimmunerkrankungen zugelassen. In vielen anderen Anwendungsgebieten sind Biopharmazeutika heute nicht mehr wegzudenken, wie Infektionskrankheiten (v. a. Impfungen), Blutgerinnungsstörungen, angeborenen Stoffwechselkrankheiten, Schlaganfall, Osteoporose oder Makuladegeneration [2]. Biologika werden zunehmend auch für Volkskrankheiten entwickelt. So wurden im Jahr 2015 z. B. die monoklonalen Antikörper Evolocumab und Alirocumab als PCSK9-Inhibitoren bei Hypercholesterinämie zugelassen [3]. Die Einführung der Checkpoint-Inhibitoren im Jahr 2011 stellte einen Meilenstein in der onkologischen Therapielandschaft dar. Seit- dem wurden weitere effektive Checkpoint-Inhibitoren, wie die PD-1-Antikörper, Nivolumab und Pembrolizumab, entwickelt und die Anwendung auf weitere Tumorentitäten ausgeweitet [4].
Mit wachsender klinischer Bedeutung und Anzahl an zugelassenen Wirkstoffen ist auch der Umsatzanteil der Biologika am Gesamtmarkt in Deutschland in den letzten Jahren stark angestiegen. Dieser hat sich seit dem Jahr 2006 fast verfünffacht und macht aktuell mit 17,1 Mrd. Euro knapp ein Drittel des pharmazeutischen Gesamtmarktes aus (Stand 2021) [5, 6]. Biologika sind inzwischen in zahlreichen Indikationen vertreten und haben in der Immunologie, Ophthalmologie, Onkologie und bei Stoffwechselerkrankungen einen überdurchschnittlich hohen Marktanteil [6].
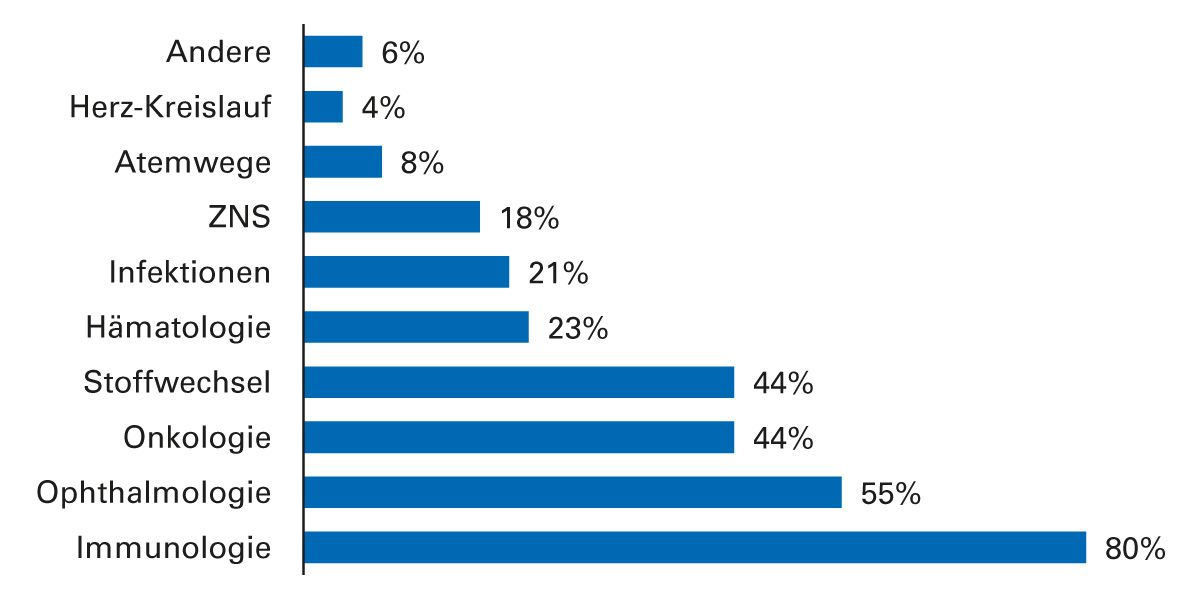
Abb. 1: Aktuelle Biologika-Marktanteile in den jeweiligen Indikationen (Stand 2021) [6]
Biologika: Definition und Herstellung
Biologika sind kein neues therapeutisches Prinzip, sondern haben sich vor allem als Impfstoffe seit über 200 Jahren in der Medizin etabliert. Im heutigen Sprachgebrauch sind Biologika jedoch definiert als hochmolekulare Wirkstoffe, die in einem biologischen Organismus mit gentechnologischen Methoden hergestellt werden [7, 8]. Die Bezeichnung Biologika hat sich für biologische Wirkstoffe (vor allem Proteine) etabliert, und dient der Abgrenzung zu niedermolekularen Wirkstoffen biologischen Ursprungs, wie z. B. Antibiotika oder Aminosäuren [8].
Definition Biologika (im heutigen Sprachgebrauch)
Biologika sind hochmolekulare Wirkstoffe, die in einem biologischen Organismus mit gentechnologischen Methoden hergestellt wurden.
Zu den Biologika gezählt werden u. a. immunologische Arzneimittel wie Antikörper, Allergene, Sera, Toxine und Impfstoffe, Gewebezubereitungen, Arzneimittel, die aus Blut und Plasma gewonnen werden, sowie Arzneimittel für neuartige Therapien („advanced therapy medicinal products“, ATMP) und andere Arzneimittel, die mittels biotechnologischer Prozesse hergestellt werden. In der Regel handelt es sich jedoch bei Biologika um Proteine, deren Komplexität wesentlich größer ist im Vergleich zu einfachen chemischen Arzneimitteln [8].
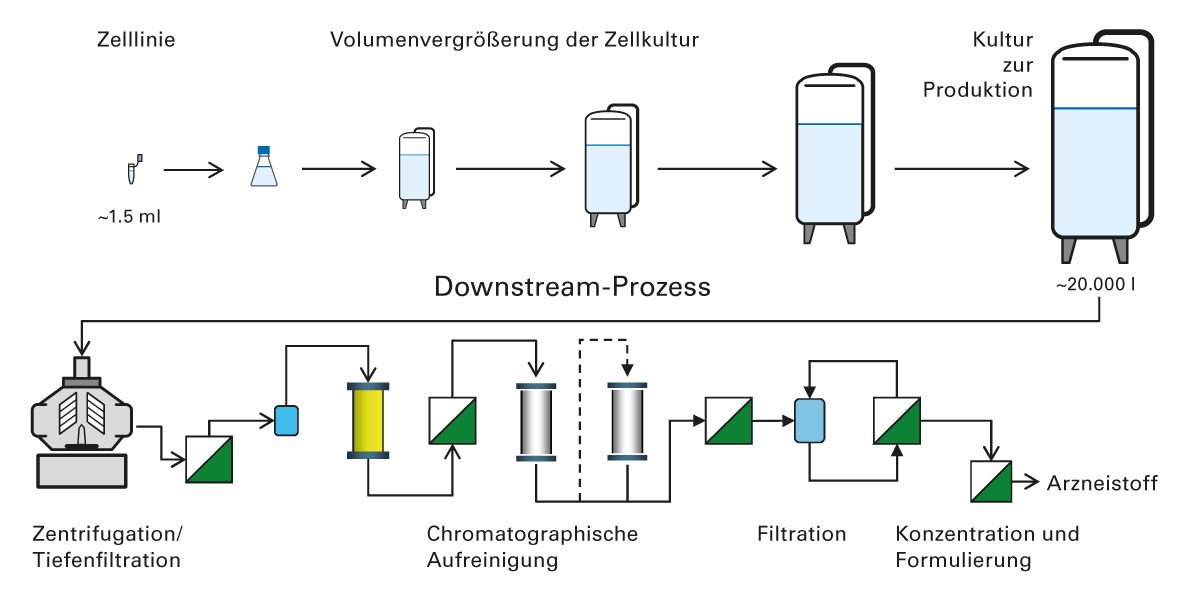
Abb. 2: Die Herstellung von Biologika ist aufwendig und umfasst mehrere Prozesse (modifiziert nach [9]).
Für die Produktion von Biologika muss ausgehend von einem (meist aus dem Menschen stammenden) Gen zunächst ein geeignetes Expressionssystem entwickelt und etabliert werden. Durch Testung unterschiedlicher Vektor-Zell-Kombinationen wird das Expressionssystem optimiert. Anschließend erfolgt die Volumenvergrößerung der Zellkultur über mehrere Schritte, bevor die eigentliche Produktion beginnen kann. In den Downstream-Prozessen wird der Wirkstoff aufgereinigt bzw. Verunreinigungen eliminiert. Dazu werden in der Regel mehrere Schritte aus Zentrifugation und Chromatogra- fie kombiniert. Zum Schluss erfolgt die stabile Formulierung, damit das Medikament für therapeutische Zwecke bei Menschen eingesetzt werden kann [9].
Mikroheterogenität ist nur innerhalb eines definierten Korridors erlaubt
Bei Biologika kommen Änderungen der Herstellungsprozesse nach der Marktzulassung häufig vor und betreffen z. B. produzierende Zelllinien, Kulturmedien oder Kulturbedingungen. Produktionsbedingte Änderungen können gewisse biochemische Eigenschaften der Proteine beeinflussen, wie z. B. Tertiärstruktur, Isoformen oder Glykosylierung. Diese Variationen werden als Mikroheterogenität bezeichnet und können Auswirkungen auf die Sicherheit und Wirksamkeit der Biologika haben. Mikroheterogenität ist ein inhärentes Kennzeichen aller Biologika und nur innerhalb eines definierten Bereichs erlaubt. Bei Änderungen des Herstellungsprozesses muss daher gewährleistet werden, dass der bei der Zulassung definierte Korridor für die Mikroheterogenität eingehalten wird [8, 10].
Die Häufigkeit von Produktionsänderungen nach der Zulassung und das damit verbundene Risiko von Auswirkungen auf Wirksamkeit und Sicherheit des Biologikums wurde in einer Studie aus dem Jahr 2016 am Beispiel von 29 monoklonalen Antikörpern (mAbs) analysiert (Abb. 3) [11]. Insgesamt wurden dabei 404 genehmigte Herstellungsänderungen identifiziert: 22 wurden als hochriskant, 286 als mäßig riskant und 96 als risikoarm eingestuft. Im Durchschnitt erfolgten 1,8 Änderungen des Produktionsprozesses pro Jahr und Biologikum. In keinem Fall hatten die Prozessänderungen jedoch klinisch relevante Auswirkungen, wie analytische und funktionelle Tests belegten [11]. Auf die inhärente Mikroheterogenität und die sich aus regelmäßigen Änderungen im Produktionsprozess ergebenden Risiken hat die EMA mit einem stringenten, wissenschaftlich basierten Zulassungs- und Überwachungsprozess nach erteilter Zulassung reagiert [12]. Für jede neue Charge eines Biologikums muss durch sensitive analytische Verfahren der Nachweis erbracht werden, dass die Unterschiede innerhalb des bei der Zulassung definierten zulässigen Korridors für Mikroheterogenität liegen [8].
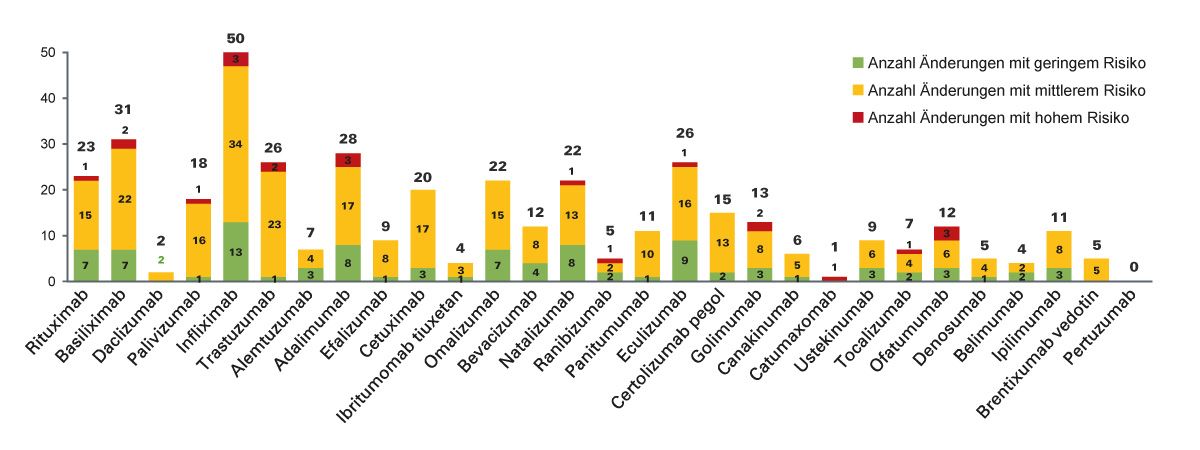
Abb. 3: Produktionsänderungen und das damit verbundene Risiko von 29 monoklonalen Antikörpern nach Marktzulassung (modifiziert nach [11])
Die EMA verfügt über beträchtliche Erfahrungen zu Prozessänderungen bei Original-Biologika, die darauf schließen lassen, dass Unterschiede im erlaubten Rahmen der Mikroheterogenität keine Auswirkungen auf die Qualität, Sicherheit und Wirksamkeit des Arzneimittels haben. Diese Erfahrungen konnte die EMA für die Definierung der regulatorischen Anforderungen an die Entwicklung von Biosimilars und deren Beurteilung im Rahmen von Zulassungsverfahren nutzen [10, 13].
Biosimilars: Definition, Entwicklung und Zulassung
Der aufwendige und zeitintensive Prozess der Entwicklung und Herstellung von Biosimilars unterscheidet sich deutlich von Generika, die als äquivalent mit ihrem Referenzprodukt gelten. Generika weisen eine einfache monomolekulare Struktur auf und können daher exakt kopiert werden. Für die Zulassung von Generika wird in der Regel nur eine Phase-1-Studie benötigt, um die Bioäquivalenz in Form einer gleichwertigen Pharmakokinetik nachzuweisen [10].
Definition Biosimilar [14]
Ein Biosimilar oder biosimilares Arzneimittel ist definiert als ein Biologikum, das eine Version des Wirkstoffs eines im europäischen Wirtschaftsraum bereits zugelassenen Biologikums (Referenzarzneimittel oder Originator) enthält.
Biosimilars dagegen sind hochkomplexe Moleküle mit aufwändigen mehrschrittigen Herstellungsprozessen, zu deren exakten Charakterisierung eine umfangreiche sensitive Analytik erforderlich ist. Für den Nachweis der Biosimilarität sind weiterhin Phase-1- und Phase-3-Studien notwendig [10]. Insgesamt dauert die Entwicklung von Biosimilars mit 8 Jahren etwa doppelt so lange gegenüber Generika. Die Kosten können bis zu 200 Mio. USD erreichen, was etwa 20 % der Entwicklungskosten des Referenzarzneimittels entspricht. Im Vergleich dazu werden zur Entwicklung von chemisch-synthetischen Generika weniger als 1 % der Entwicklungskosten des Original-Arzneimittels benötigt [15].
Für die pharmazeutische Qualität von Biosimilars gelten dieselben hohen Standards wie für jedes neue biologische Arzneimittel. Die EMA gibt die analytischen Methoden zur physikochemischen Charakterisierung, Bestimmung der biologischen Aktivität, der Reinheit und der Spezifikationen des Biosimilars vor [14].
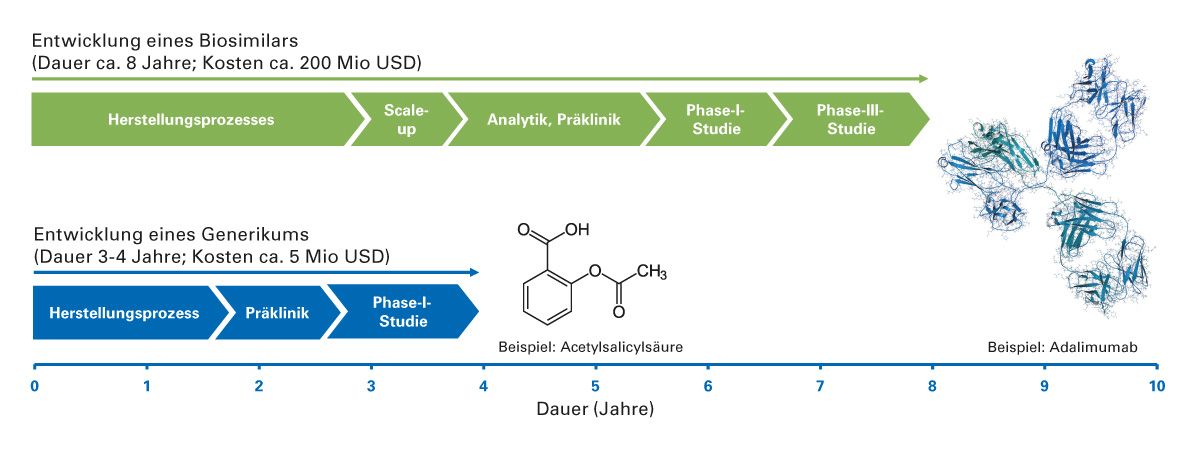
Abb. 4: Die Entwicklung von Biosimilars ist deutlich zeit- und kostenintensiver im Vergleich zu Generika (Angaben nach [15]).
Generell wird für biosimilare Proteine gefordert, dass sie die gleiche Aminosäurensequenz, die gleiche Proteinfaltung und die gleiche biologische Aktivität wie das Referenzarzneimittel aufweisen müssen. Posttranslationale Modifikationen bzw. Mikroheterogenität werden nur in dem zulässigen Umfang akzeptiert wie auch beim Referenzarzneimittel. Dieser wird so definiert, dass er nicht klinisch relevant ist, also weder die Immunogenität erhöht noch die Wirksamkeit beeinträchtigt [8].
Extrapolation: ein wissenschaftlich anerkanntes Prinzip
Ist ein Biosimilar einem Referenzarzneimittel sehr ähnlich und weist eine vergleichbare Sicherheit und Wirksamkeit bei einer therapeutischen Indikation auf, können die Sicherheits- und Wirksamkeitsdaten gegebenenfalls auf andere zugelassene Indikationen des Referenzarzneimittels extrapoliert werden [8, 10].
Der Nachweis der vergleichbaren Wirksamkeit und Sicherheit erfolgt dabei in einer Phase-3-Studie für eine sensitive Indikation. Die Extrapolation ist keine Zulassungsvereinfachung für Biosimilars, sondern ein wissenschaftlich begründeter Prozess zur Qualitätssicherung biologischer Referenzarzneimittel bei Produktionsänderungen, der auch auf Biosimilars angewendet wird [8, 16]. Das bedeutet, dass für bestimmte Indikationen weniger klinische Studien oder keine Studien mit dem Biosimilar durchgeführt werden müssen. Die Extrapolation der Daten auf andere Indikationen muss aber immer durch umfangreiche wissenschaftliche Evidenz gestützt werden, die in robusten Vergleichbarkeitsstudien generiert wurden [8, 10, 16].
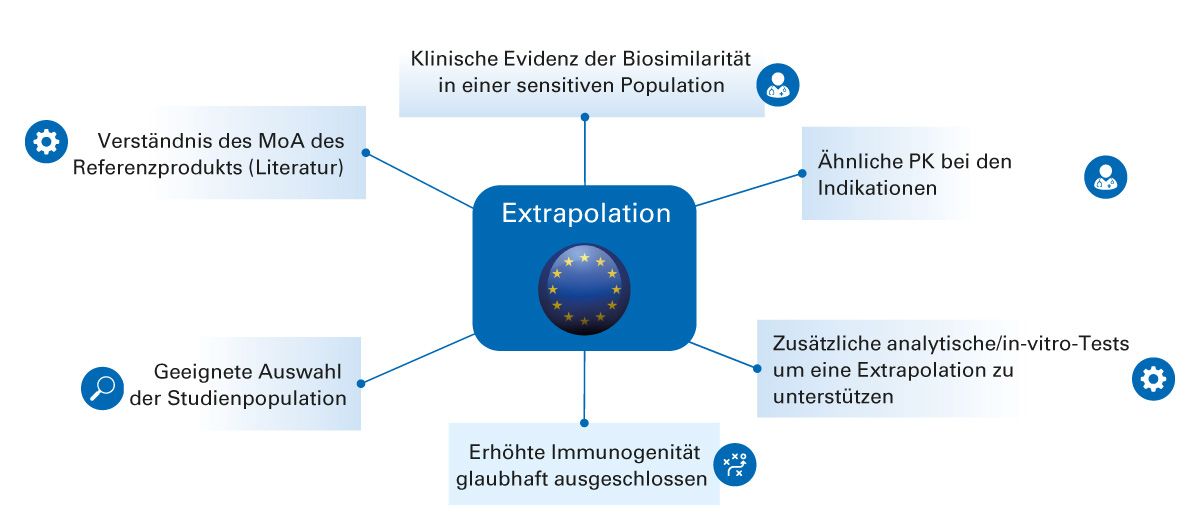
Abb. 5: EMA-Anforderungen an die Extrapolation (Abbildung modifiziert nach [16])
PK: Pharmakokinetik, MoA: Wirkmechanismus
Nur wenn eine mit dem Referenzarzneimittel vergleichbare klinische Ähnlichkeit und Wirksamkeit in einer der zugelassenen Indikationen des Referenzarzneimittels nachgewiesen wurde und keine klinisch relevanten Unterschiede bestehen, verzichtet die EMA für die weiteren Indikationen auf zusätzliche Studien, wenn der relevante Wirkmechanismus bzw. die an den extrapolierten Indikationen beteiligten Rezeptoren die gleichen sind. Die Anforderungen der EMA an die Evidenz als Voraussetzung für die Extrapolation sind in Abbildung 5 dargestellt [16].
Biosimilars: Bedeutung, Indikationen und Ausblick
Im Jahr 2006 kam das erste Biosimilar zu Somatropin nach Patentablauf des Referenzarzneimittels auf den Markt. Seitdem erhielten immer mehr Biosimilars die Zulassung in der EU und in den USA. Bis zum Jahr 2022 wurden 86 Biosimilars in der EU zugelassen und überprüft [17]. Die EMA verfügt damit über eine mehr als 15-jährige Erfahrung in der Bewertung, Überwachung und Pharmakovigilanz von Biosimilars [17, 18].
Durch die Einführung der Biosimilars in den Markt wurde ein Wettbewerb ausgelöst, der sinkende Preise zur Folge hatte [19, 20]. Der zunehmende Wettbewerb führte auch zur Preissenkung der Originalpräparate. Weitere Kosteneinsparungen wurden durch Rabattverträge und Festbeträge erreicht. Zusätzlich sank nach Markteintritt eines Biosimilars auch das Preisniveau der gesamten ATC (Anatomisch-Therapeutisch-Chemisches Klassifikationssystem)-Gruppe, d. h. also auch die Preise von Biologika, für die es noch kein Biosimilar gibt [15, 21]. Beispielsweise halbierte sich der Preis des TNFα- Inhibitors Adalimumab seit Einführung der Biosimilars von rund 61 Euro (im Jahr 2018) auf 34 Euro pro Tagesdosis (Stand 2021) [22]. Insgesamt wurden durch Biosimilars seit 2011 bereits mehr als 4 Milliarden Euro im deutschen Gesundheitssystem eingespart (Abb. 6) [23].
Die durch den Wettbewerb der Biosimilars sinkenden Therapiekosten entlasten die gesetzlichen Krankenkassen und verbessern die Versorgung der Patienten. Mehr Betrof- fene erhalten einen schnelleren Zugang zu einer leitliniengerechten Biologika-Therapie, wie eine aktuelle Analyse zur Versorgung von Rheuma-Patienten in Deutschland zeigte [24]. Demnach musste ein Rheuma-Patient im Jahr 2014 (vor Einführung der Biosimilars) noch durchschnittlich 3,2 Jahre auf eine Biologika-Therapie warten. Dagegen waren es im Jahr 2019 nur noch 2,2 Jahre [24]. Gleichzeitig stieg der Anteil an Rheuma-Patienten mit einer Biologika-Therapie von 12,3 % im Jahr 2014 auf 20,4 % im Jahr 2019 an, was nicht nur durch den wissenschaftlichen Fortschritt, sondern auch durch die bessere Versorgung erklärt werden kann [24].
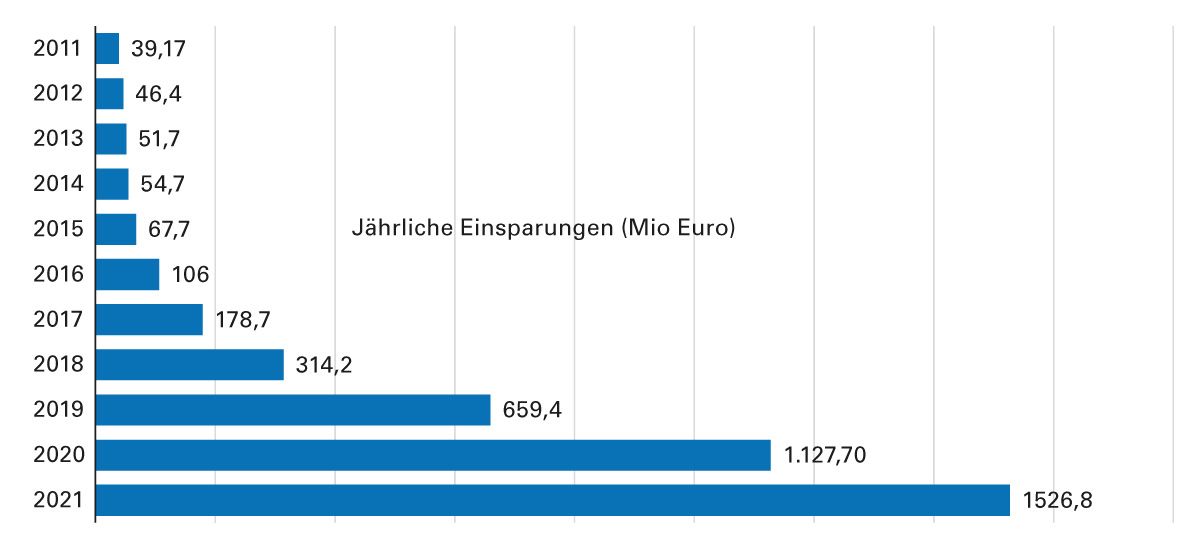
Abb. 6: Jährliche Kosteneinsparungen durch Biosimilars in Deutschland seit 2011 (modifiziert nach [23])
Sicherheit und Austauschbarkeit von Biosimilars
Obwohl EU-Biosimilars eine ausgezeichnete Sicherheitsbilanz vorweisen, sind viele Ärzte immer noch skeptisch und zögern bei der Umstellung der Patienten von Referenzprodukten auf Biosimilars [25].
Um die Bedenken der Kliniker zu adressieren, wurde eine umfassende Datenanalyse durchgeführt [18]. Dazu wurden alle verfügbaren Dokumente der EMA zur Sicherheit, Immunogenität und Austauschbarkeit von Biosimilars bis zum Stichtag am 31.07.2020 analysiert und im darauffolgenden Jahr durch Kurki et al. publiziert [26]. Bei den EMA- Dokumenten handelte es sich um EPAR (European Public Assessment Reports), PSUR (Periodic Safety Update Reports) [27] und PSUSA (Periodic Safety Update Single Assessments) [18].
Die umfassende Datenanalyse von Kurki et al. ergab, dass alle Biosimilars ein vergleichbares Sicherheitsprofil zu den Referenzprodukten aufwiesen ein- schließlich der Immunogenität [26]. Trotz einer beachtlichen Exposition von über 1 Millionen Patientenjahren wurden keine Sicherheitsbedenken identifiziert. Da die jüngsten Zulassungen für Biosimilars eine subkutane Selbstverabreichung durch Patienten erlauben, wurden auch Selbstverabreichungsformen, wie Fertigspritzen und Pens, untersucht. Die unterschiedlichen Selbstverabreichungsformen der Biosimilars erhöhten jedoch nicht das Risiko für unerwünschte Ereignisse [26].
Neben dieser umfassenden Datenanalyse wurden auch bei der Überprüfung von 178 Switch-Studien keine Sicherheitsbedenken identifiziert [28-31]. Der Wechsel von einem Referenzprodukt auf ein Biosimilar oder von einem Biosimilar auf ein anderes Biosimilar verursachte keine unerwünschten Ereignisse oder einen Wirksamkeitsverlust. Auch die Immunogenität wurde durch einen Switch nicht erhöht oder induziert [28-31].
Insgesamt wurde das Konzept der Biosimilarität bestätigt und gezeigt, dass die Austauschbarkeit von EU-zugelassenen Biosimilars gegeben ist. Weitere systematische Switch-Studien sind nach Ansicht der Autoren nicht erforderlich [26].
EMA bestätigt das Konzept der Biosimilarität
Auf Basis aller vorhandenen Daten bestätigten EMA und HMA (Heads of Medicine Agencies) in einer gemeinsamen Stellungnahme vom 19.09.2022 das Konzept der Biosimilarität, die Austauschbarkeit und Sicherheit der Biosimilars und schafften somit Klarheit für Ärzte und Patienten [32]. Die nationalen Arzneimittelbehörden der EU-Mitgliedstaaten haben in den letzten 15 Jahren insgesamt 86 zugelassene Biosimilar-Arzneimittel im Ausschuss für Humanarzneimittel CHMP bei der EMA geprüft und nach der Zulassung überwacht. Dabei zeigte sich, dass EU-Biosimilars in den vergangenen 15 Jahren eine ausgezeichnete Sicherheitsbilanz vorweisen [17, 18].
Vorerst keine Aut-idem-Substitution von Biosimilars in Deutschland (Stand 2022)
Trotz der eindeutig positiven Stellungnahme der EMA und HMA zur Austauschbarkeit von EU-zugelassenen Biosimilars mit Referenzprodukten und anderen Biosimilars gibt es in Deutschland vorerst keine automatische Substitution [33]. Die Aut-idem-Substitution von Biosimilars ist EU-weit unterschiedlich geregelt, aber in vielen Mitgliedsstaaten wird der automatische Austausch von Original-Biologika mit ihren Biosimilars bereits praktiziert. Laut dem Gesetz für mehr Sicherheit in der Arzneimittelversorgung (GSAV) aus dem Jahr 2016 war in Deutschland die automatische Substitution von Biologika in der Apotheke ab dem 16. August 2022 vorgesehen – ähnlich der Vorschriften für den Austausch bei Generika. Voraussetzung ist, dass der Gemeinsame Bundesausschuss (G-BA) vorab eine Austauschbarkeit in Bezug auf ein biologisches Referenzarzneimittel festgestellt hat. Laut Regierungsentwurf zum GKV-Finanzstabilisierungsgesetz wurde jedoch die Frist um ein Jahr auf August 2023 verlängert. Begründet wurde dies mit erheblichen Einwänden von Sachverständigen der medizinischen Wissenschaft und Praxis, Spitzenorganisationen der pharmazeutischen Unternehmer, betroffenen pharmazeutischen Unternehmen und Berufsvertretungen der Apotheker [33].
Zahlreiche Neuzulassungen von Biosimilars in den nächsten Jahren
Derzeit laufen etwa 300 Zulassungsstudien von Biosimilars und bis 2030 werden 139 Biologika von Patentabläufen betroffen sein [34, 35]. Wichtige Biosimilar-Markteinführungen werden insbesondere in der Onkologie, Ophthalmologie sowie zur Osteoporose-Behandlung erwartet. In den nächsten Jahren wird mit weiteren jährlichen Kosteneinsparungen durch Biosimilars in einer Höhe von 700 Mio. € bis 1,2 Mrd. € für die gesetzlichen Krankenversicherungen in Deutschland gerechnet [36].
Biosimilars: Erfahrungen aus der Praxis
Seit der Erstzulassung 2006 wurde eine wachsende Anzahl an Switch-Studien in unterschiedlichen Indikationen durchgeführt, die sämtlich die Sicherheit und Austauschbarkeit von Biosimilars in der klinischen Praxis bestätigten [8, 17, 26, 28-32]. Bei keinem einzigen Biosimilar traten unbekannte Sicherheitsprobleme, wie z. B. schwere Nebenwirkungen auf. Es wurden keine Unterschiede hinsichtlich Wirksamkeit oder der Art, Häufigkeit und Schweregrad von Nebenwirkungen beim Wechsel auf ein Biosimilar beobachtet [8].
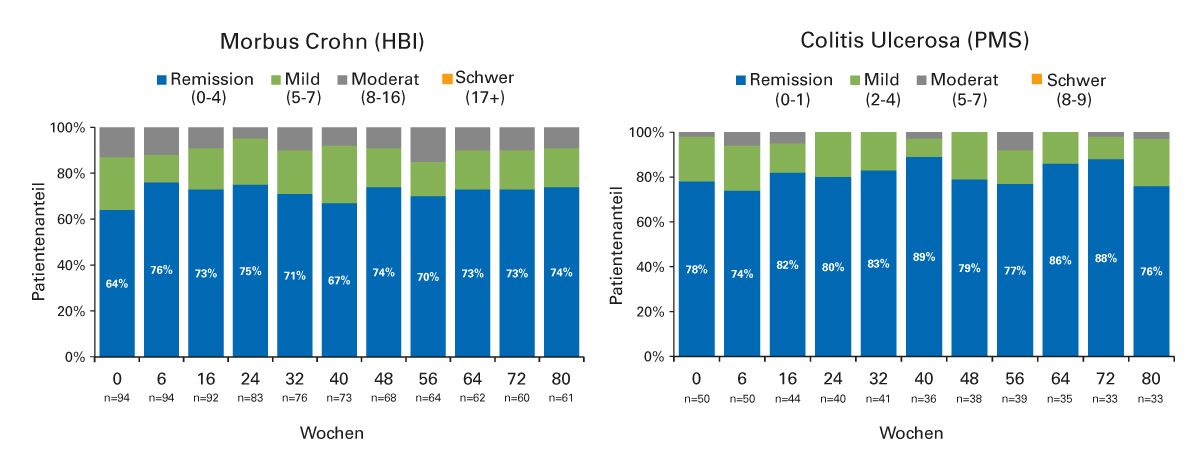
Abb. 7: Patienten mit Remission nach Umstellung auf das Infliximab-Biosimilar SB2 (modifiziert nach [37]) HBI: Harvey-Bradshaw-Index; PMS: partieller Mayo-Score
Exemplarisch wird hier eine Switch-Studie vorgestellt, in der Patienten mit chronisch- entzündlichen Darmkrankheiten vom Infliximab-Originalpräparat auf das Biosimilar SB2 umgestellt wurden (Abb. 7) [37]. In der prospektiven monozentrischen Studie wurde die Krankheitsaktivität von 94 Patienten mit Morbus Crohn mittels Harvey-Bradshaw-Index (HBI) und von 50 Patienten mit Colitis ulcerosa mittels partiellem Mayo-Score (PMS) erfasst. Über die gesamte Beobachtungsdauer von insgesamt 80 Wochen blieb der Patientenanteil in Remission konstant. Auch die CRP-Werte blieben im Normbereich. Insgesamt wurde keine Änderung der Wirksamkeit oder Immunogenität nach Switch auf das Infliximab-Biosimilar beobachtet und der Wechsel wurde gut vertragen [37].
Nocebo-Effekte durch positives Framing minimieren
Als Nocebo-Effekte werden negative Reaktionen bezeichnet, einschließlich neuer oder sich verschlechternder Symptome und Nebenwirkungen, die nicht auf die pharmakologische Wirkung der Behandlung selbst zurückzuführen sind [8, 38, 39]. Es handelt sich dabei um unerklärbare Effekte, die auf Basis von negativen Erwartungen des Patienten über die schädlichen Auswirkungen einer Therapie entstehen. Darüber hinaus gehören falsche Zuordnung der Symptome, vorherige Lernphasen, Kontextfaktoren und Eigenschaften der Behandlung, aber auch verbale und nonverbale Kommunikation der Behandler und Patient-Behandler-Interaktionen zu den Pathomechanismen des Nocebo- Effekts [38].
Wissenslücken und Verständnisprobleme von Patienten und Ärzten sowie Fehleinschätzungen über Arzneimittel können zu Unsicherheiten und negativen Bewertungen von neuartigen Therapien beitragen. Nachweislich können sich Nocebo-Effekte negativ auf Adhärenz, Nebenwirkungen, Symptomlinderung und Abbruchraten der medikamentösen Therapie auswirken [8, 38]. So ergab eine Meta-Analyse von mehr als 30 Studien mit über 3.000 Patienten, dass die mittlere Abbruchrate nach Switch auf ein Biosimilar in offenen Studien mit 14 % doppelt so hoch lag gegenüber der Abbruchrate in doppelblinden Studien mit 7 % [40]. Laut Umfragen sind negative Ansichten über Biosimilars und Generika nicht nur bei Patienten weit verbreitet. Etwa 20–30 % der Apotheker und Ärzte sehen Generika und Biosimilars als weniger effektiv und qualitativ schlechter an als die jeweiligen Markenprodukte [38].
Bei einer Umstellung der Patienten von Referenzarzneimitteln auf Biosimilars können Nocebo-Effekte die Adhärenz und den therapeutischen Erfolg gefährden. Ausführliche Informationen und Beratungen der Patienten sind geeignete Gegenmaßnahmen, um Nocebo-Effekte zu minimieren. Weiterhin ist eine gute Kommunikation zwischen Arzt und Patient ein wichtiger Schritt, um das Vertrauen des Patienten in ein Biosimilar aufzubauen. Eine positive Haltung und Sprache (Framing) können sich förderlich auf die Adhärenz auswirken [41]. Eine neuere Studie belegt, dass positives Framing im Vergleich zu einer Standardrisikoinformation die Nebenwirkungen nach der Einnahme eines Placebos um 34 % reduzierte [40]. Relevante Informationen zur Definition und Zulassung von Biosimilars sowie zu Wirksamkeit und Sicherheit können die Bedenken und Ängste der Patienten abbauen. Dabei sind präzise Antworten in einer patientenverständlichen Sprache besonders wichtig [8, 42]. Einige Beispiele für eine Nocebo-induzierende und -limitierende Kommunikation zeigt Tabelle 1 [8].
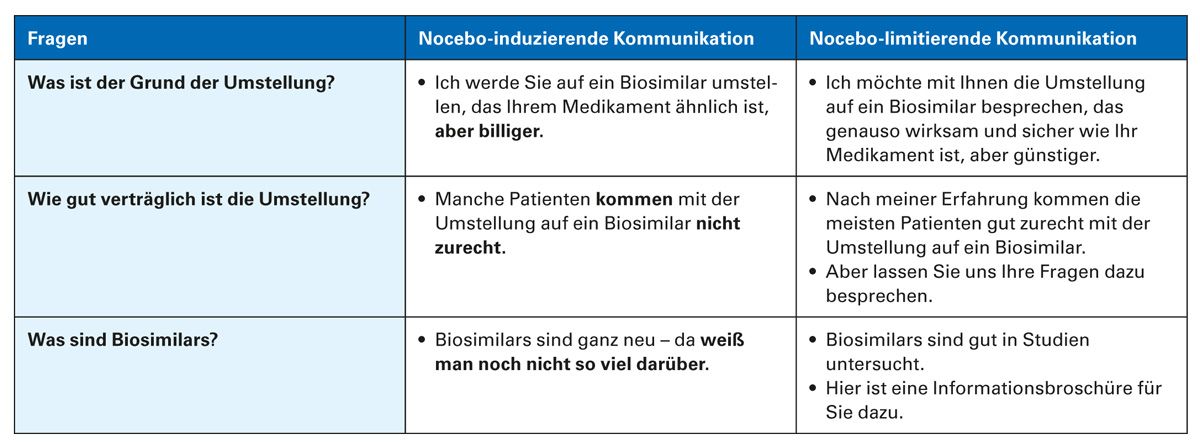
Tab. 1: Beispiele für eine Nocebo-induzierende und Nocebo-limitierende Arzt-Patienten-Kommunikation (modifiziert nach [8])
Fazit
Biologika sind hochkomplexe Moleküle, die mit Hilfe von lebenden Organismen hergestellt werden. Mikroheterogenität als Kennzeichen aller Biologika ist nur im bestimmten Korridor erlaubt und muss bei jeder Charge bzw. bei Änderungen im Herstellungsprozess neu überprüft werden. Die EMA nutzte diese langjährigen, umfangreichen Erfahrungen, um Regularien für die Zulassung von Biosimilars zu definieren.
Bei der Entwicklung von Biosimilars umfassen die regulatorischen Anforderungen u. a. den Nachweis der Bioäquivalenz und einer vergleichbaren Wirksamkeit, Sicherheit und Immunogenität durch Phase-1- und Phase-3-Studien. Unter bestimmten Voraussetzungen und insbesondere bei ausreichender wissenschaftlicher Evidenz ist die Extrapolation erlaubt, d. h. eine Phase-3-Studie erfolgt in einer sensitiven Indikation und die Daten können auf die übrigen zugelassenen Indikationen des Referenzproduktes extrapoliert werden. Die Extrapolation ist keine Sonderregel für die erleichterte Zulassung von Biosimilars, sondern ein allgemein anerkanntes wissenschaftliches Prinzip. Seit 2006 wurden in der EU 86 Biosimilars zugelassen (Stand 2022). Dies hat zu Kosteneinsparungen in Milliardenhöhe geführt. Bis 2030 laufen 139 Patente von Biologika aus, daher werden zahlreiche weitere Neuzulassungen von Biosimilars erwartet.
Die klinischen Erfahrungen über 15 Jahre haben gezeigt, dass EU-zugelassene Biosimilars mit Referenzprodukten und anderen Biosimilars austauschbar sind. Umfassende Analysen von Sicherheitsdaten sowie 178 Switch-Studien bestätigten das Konzept der Biosimilarität und das Prinzip der Extrapolation. Für die Umstellung von Biologika auf Biosimilars und für die Therapie mit Biosimilars wurden keinerlei Sicherheitsbedenken identifiziert. Die EMA bestätigte daher die Austauschbarkeit der Biosimilars und schaffte somit Klarheit für Ärzte und Patienten.
Quellen
1. Vecchio I, Tornali C, Bragazzi NL et al. The Discovery of Insulin: An Important Milestone in the History of Medicine. Front Endocrinol (Lausanne). 2018; 9:613.
2. Lücke J, Bädecker M, and Hildinger M. 10. Biotech-Report. 2015; Available from: https://www.vfa-bio.de/vb-de/ vb-presse/vb-publikationen.
3. Blanchard V, Khantalin I, Ramin-Mangata S et al. PCSK9: from biology to clinical applications. Pathology. 2019; 51(2):177-183.
4. Lamos C and Hunger RE. [Checkpoint inhibitors-indications and application in melanoma patients]. Z Rheumatol. 2020; 79(8):818-825.
5. Wartenberg F. IQVIA Fokus Biosimilars 2022.
6. Lücke J, Bädecker M, and Hildinger M. Medizinische Biotechnologie in Deutschland 2022. 2022; Available from: www. vfa-bio.de/publikationen
7. Freissmuth M, Pharmakologie & Toxikologie. 2020, Heidelberg: Springer Medizin.
8. Dicheva S, Bräutigam K, and Ludwig W-D. Biosimilars. Leitfaden der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ). 2021.
9. Vulto AG and Jaquez OA. The process defines the product: what really matters in biosimilar design and production? Rheumatology (Oxford). 2017; 56(suppl_4):iv14-iv29.
10. European Medicines Agency. Biosimilars in der EU. Leitfaden für medizinische Fachkräfte. 2019; Available from: ht- tps://www.ema.europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_de.pdf.
11. Vezér B, Buzás Z, Sebeszta M et al. Authorized manufacturing changes for therapeutic monoclonal antibodies (mAbs) in European Public Assessment Report (EPAR) documents. Curr Med Res Opin. 2016; 32(5):829-34.
12. European Medicines Agency. Similar biological medicinal products containing biotechnology-derived proteins as active substance: non-clinical and clinical issues. EMEA/CHMP/BMWP/42832/2005 Rev. 1. 2014; Available from: https:// www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing- biotechnology-derived-proteins-active_en-2.pdf.
13. Wolff-Holz E, Müller-Berghaus J, and Weise M. Biosimilars in der Onkologie (Teil 1): Dank strenger Regulierung keine zweite Wahl. Dtsch Arztebl International. 2019; 116:35-36.
14. European Medicines Agency. Guideline on similar biological medicinal products. 2014; Available from: https://www. ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf.
15. Bauer C, May U, and Giulini-Limbach C. Bedarfsgerechte Versorgung mit modernen Biopharmazeutika nach 2022. 2022; Available from: https://www.monitor-versorgungsforschung.de/efirst/Versorgung_Bios_2022.
16. Weise M, Kurki P, Wolff-Holz E et al. Biosimilars: the science of extrapolation. Blood. 2014; 124(22):3191-6.
17. Paul-Ehrlich-Institut. Gemeinsame Stellungnahme der EMA und HMA zur Austauschbarkeit von Biosimilars mit Referenzarzneimittel und gleichwertigen Biosimilars. Available from: https://www.pei.de/DE/newsroom/hp- meldungen/2022/220923-ema-hma-stellungnahme-biosimilar-austauschbarkeit.html.
18. Wolff-Holz E. Sicherheit, Immunogenität und Austauschbarkeit von Biosimilars – monoklonale Antikörper und Fusionsproteine mit Antikörperanteil im Fokus. 2021; Available from: https://www.pei.de/SharedDocs/Downloads/ DE/newsroom/bulletin-arzneimittelsicherheit/einzelartikel/2022-biosimilars.pdf? blob=publicationFile&v=4.
19. Hübel K, Kron F, and Lux MP. Biosimilars in oncology: Effects on economy and therapeutic innovations. Eur J Cancer. 2020; 139:10-19.
20. Vogler S, Schneider P, Zuba M et al. Policies to Encourage the Use of Biosimilars in European Countries and Their Potential Impact on Pharmaceutical Expenditure. Front Pharmacol. 2021; 12:625296.
21. Morf H and Witte T. [Use of biosimilars in the treatment of rheumatoid arthritis : An overview]. Z Rheumatol. 2022; 81(2):110-117.
22. Probiosimilars. 2022; Available from: https://probiosimilars.de/themen/die-bedeutung-von-biosimilars/ bedeutung-fuer-das-gesundheitssystem/.
23. Probiosimilars. Biosimilars in Zahlren. 2021; Available from: https://probiosimilars.de/app/uploads/2022/07/ Biosimilars-in-Zahlen_Jahr-2021.pdf.
24. Hörbrand F, Schuch F, Bleß H-H et al. PHARAO-Studie: Arzneimittelversorgung entzündlich rheumatischer Erkrankun- gen. Zeitschrift für Rheumatologie. 2022.
25. Sarnola K, Merikoski M, Jyrkkä J et al. Physicians‘ perceptions of the uptake of biosimilars: a systematic review. BMJ Open. 2020; 10(5):e034183.
26. Kurki P, Barry S, Bourges I et al. Safety, Immunogenicity and Interchangeability of Biosimilar Monoclonal Antibodies and Fusion Proteins: A Regulatory Perspective. Drugs. 2021; 81(16):1881-1896.
27. European Medicines Agency. European public assessment reports: background and context. 2022; Available from: https://www.ema.europa.eu/en/medicines/what-we-publish-when/ european-public-assessment-reports-background-context.
28. Mckinnon RA, Cook M, Liauw W et al. Biosimilarity and Interchangeability: Principles and Evidence: A Systematic Review. BioDrugs. 2018; 32(1):27-52.
29. Cohen HP, Blauvelt A, Rifkin RM et al. Switching Reference Medicines to Biosimilars: A Systematic Literature Review of Clinical Outcomes. Drugs. 2018; 78(4):463-478.
30. Barbier L, Ebbers HC, Declerck P et al. The Efficacy, Safety, and Immunogenicity of Switching Between Reference Biopharmaceuticals and Biosimilars: A Systematic Review. Clin Pharmacol Ther. 2020; 108(4):734-755.
31. Wiland P, Batko B, Brzosko M et al. Biosimilar switching – current state of knowledge. Reumatologia. 2018; 56(4):234-242.
32. European Medicines Agency. Statement on the scientific rationale supporting interchangeability of biosimilar medicines in the EU. 2022; Available from: https://www.ema.europa.eu/en/documents/public-statement/ statement-scientific-rationale-supporting-interchangeability-biosimilar-medicines-eu_en.pdf.
33. Ärztezeitung. EMA: Zugelassene Biosimilars sind austauschbar mit Referenz-Arzneimittel. 2022; Available from: https://www.aerztezeitung.de/Wirtschaft/EMA-Zugelassene-Biosimilars-sind-austauschbar-mit-Referenz-Arzneimit- tel-432683.html.
34. IQVIA. Fokus Biosimilars. 2022; Available from: https://www.iqvia.com/-/media/iqvia/pdfs/germany/publications/ fokus-biosmilars/newsletter-fokus-biosimilars-ausgabe-10.pdf.
35. Probiosimilars. Biosimilar-Produktion weltweit: So stark ist der Standort Deutschland! 2022.
36. Schmitt N and Heltweg B. Biosimilars im Fokus. 2022; Available from: https://www.barmer.de/resource/blob/1127018
/40260c8b306412a4c1f8236b782e9297/bifg-epaper-biosimilars-im-fokus-data.pdf.
37. Fischer S, Cohnen S, Klenske E et al. Long-term effectiveness, safety and immunogenicity of the biosimilar SB2 in inflammatory bowel disease patients after switching from originator infliximab. Therap Adv Gastroenterol. 2021; 14:1756284820982802.
38. Braun J, Tsiami S, Buehring B et al. Biosimilars und der Nocebo-Effekt. rheuma plus. 2020; 19:179–189.
39. Petrie KJ and Rief W. Psychobiological Mechanisms of Placebo and Nocebo Effects: Pathways to Improve Treatments and Reduce Side Effects. Annu Rev Psychol. 2019; 70:599-625.
40. Odinet JS, Day CE, Cruz JL et al. The Biosimilar Nocebo Effect? A Systematic Review of Double-Blinded Versus Open- Label Studies. J Manag Care Spec Pharm. 2018; 24(10):952-959.
41. Colloca L and Barsky AJ. Placebo and Nocebo Effects. N Engl J Med. 2020; 382(6):554-561.
42. Hansen E, Zech N, and S. B. Nocebo, Aufklärung und Arzt-Patienten-Kommunikation. Nervenarzt. 2020; 91:691-699.
Bildquellen
Titel: © ustas – stock.adobe.com
Abb 4: © molekuul.be – stock.adobe.com
Tutorielle Unterstützung
Die tutorielle Unterstützung der Fortbildungsteilnehmer erfolgt durch unseren ärztlichen Leiter Dr. med. Alexander Voigt in Zusammenarbeit mit der arztCME-Redaktion. Inhaltliche Fragen können über das Kommentarfeld, direkt per Mail an service@arztcme.de oder via Telefon unter Tel.: +49(0)180-3000759 gestellt werden. Inhaltliche Fragen werden von unserem ärztlichen Leiter bzw. nach Rücksprache mit diesem und evtl. dem Autor auch von der arztCME-Redaktion beantwortet.
Technischer Support
Der technische Support der arztCME-Online-Akademie erfolgt durch geschulte Mitarbeiterinnen und Mitarbeiter des Betreibers health&media GmbH unter der E-Mail-Adresse technik@arztcme.de oder via Telefon unter Tel.: 49(0)180-3000759.