Einsatz von Anti-TNF-Biosimilars – Erfahrungen und Hinweise für die Praxis
Interessengebiete: Allgemeinmedizin und Innere Medizin, Haut- und Geschlechtskrankheiten, Gastroenterologie, Rheumatologie
Die Einführung von Biosimilars bietet ökonomische Vorteile für das Gesundheitssystem. Ärzte müssen selbst gut über Biosimilars informiert sein, auch um gegenüber dem Patienten die Verordnung von Biosimilars überzeugend vertreten zu können und die Akzeptanz für deren Einsatz zu steigern.
Diese Fortbildung soll Ärzte über Definition und Zulassung von Biosimilars sowie zu Wirksamkeit und Sicherheitsprofil informieren. Der Fokus liegt dabei auf Tumornekrosefaktor(TNF)-Inhibitoren, welche fachübergreifend in der Rheumatologie, Dermatologie und Gastroenterologie eingesetzt werden.
Kursinhalt
Inhaltsverzeichnis
- Einleitung
- TNF-Inhibitoren – seit über 20 Jahren bewährt
- Mono- vs. Kombinationstherapie von TNF- Inhibitoren mit Immunmodulatoren
- Strategien bei Therapieversagen unter TNF-Inhibitor-Therapie
- Biosimilars – Äquivalente Wirksamkeit und Sicherheitsprofil zum Referenzprodukt
- Mikroheterogenität als Merkmal aller biologischen Arzneimittel
- Klinische Vergleichsstudien zu Infliximab, Adalimumab und Etanercept
- Extrapolation der Zulassung auf andere Indikationen
- Biosimilars bringen ökonomische Vorteile für das Gesundheitssystem
- Wirksamkeit und Sicherheitsprofil von Biosimilars
- Real-World-Daten – Erfahrungen aus der klinischen Praxis
- Mehrfacher Switch – kein Einfluss auf Wirksamkeit, Sicherheitsprofil und Immunogenität
- Nocebo-Effekt als Grund für vorzeitige Therapieabbrüche
- Automatische Substitution von Biologika
- Fazit
- Literatur
Einleitung
Diese CME richtet sich an Rheumatologen, Dermatologen, Gastroenterologen und Internisten. Hochwirksame Biologika haben sich in den letzten 25 Jahren in der Therapie von Tumor- und Autoimmunerkrankungen etabliert. Die Herstellung ist jedoch mit hohen Kosten und einer eingeschränkten Verfügbarkeit in weiten Teilen der Welt verbunden. Die Einführung von Biosimilars hat maßgeblich zur Preissenkung beigetragen und bietet ökonomische Vorteile für das Gesundheitssystem. Eine finanzielle Entlastung ist jedoch nur realisierbar, wenn Biosimilars durch die verordnenden Ärzte und die Patienten akzeptiert werden. Um gegenüber dem Patienten die Verordnung von Biosimilars überzeugend vertreten zu können, müssen Ärzte selbst gut über Biosimilars informiert sein. Mit diesem Ziel bietet diese CME relevante Informationen zur Definition und Zulassung von Biosimilars sowie zu Wirksamkeit und Sicherheitsprofil. Der Fokus liegt dabei auf Tumornekrosefaktor-(TNF-)Inhibitoren, welche fachübergreifend in der Rheumatologie, Dermatologie und Gastroenterologie eingesetzt werden.
TNF-Inhibitoren – seit über 20 Jahren bewährt
Der Tumornekrosefaktor (TNF) wurde erstmals 1975 als Mediator für Endotoxin-induzierte Tumornekrose beschrieben [1] und gehört zu den Zytokinen. Er wird hauptsächlich von Makrophagen ausgeschüttet und ist als proinflammatorischer multifunktionaler Signalstoff des Immunsystems grundlegend an vielen Prozessen bei der Entzündung und Abwehr intrazellulärer Krankheitserreger beteiligt (Abbildung 1).
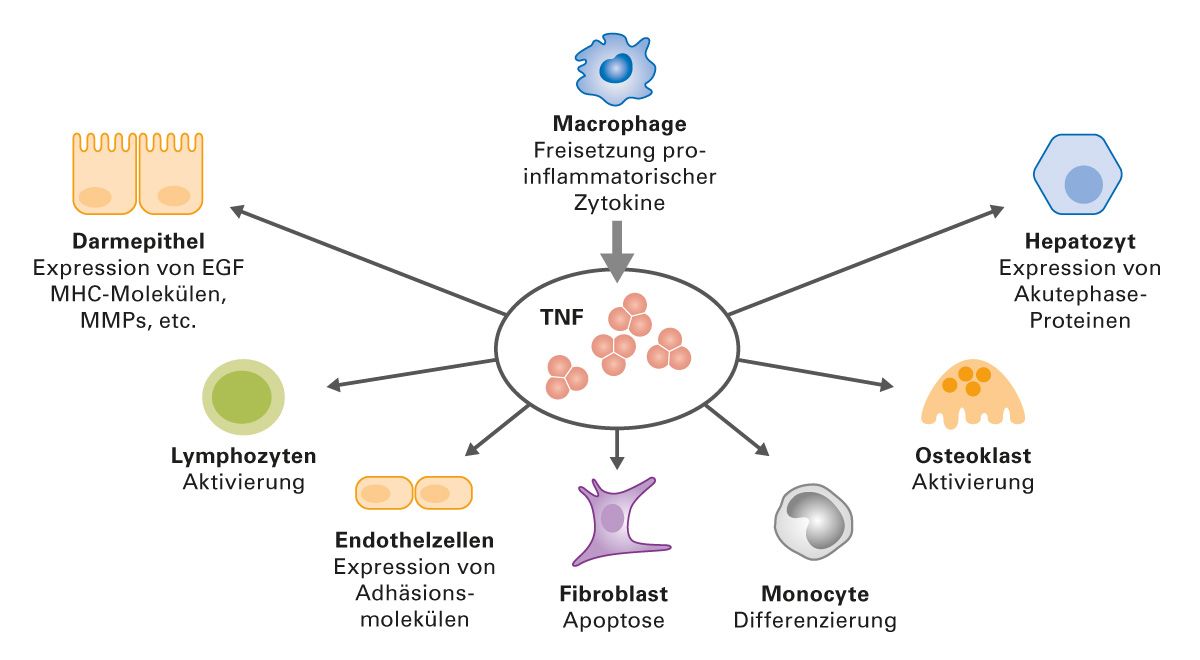
Abb. 1: Biologische Wirkungen des TNF
EGF, Epidermaler Wachstumsfaktor; MHC, Haupthistokompatibilitätskomplex; MMP, Matrix-Metalloprotease; TNF, Tumornekrosefaktor. Modifiziert nach 1. Korzenik JR, Podolsky DK (2006); 2. Esposito E, Cuzzocrea S (2011)
Infolge der durch TNF ausgelösten Entzündungsprozesse kann es zu immunvermittelten entzündlichen Erkrankungen kommen, wie rheumatoide Arthritis (RA), Psoriasis (Pso), axiale Spondyloarthritis, Psoriasis-Arthritis (PsA) oder den chronisch entzündlichen Darmerkrankungen (CED) Morbus Crohn (MC) und Colitis ulcerosa (CU). Mit dem Verständnis der jeweiligen krankheitsspezifischen Immunpathogenese hat sich die Blockade des zentralen Entzündungsfaktors TNF durch TNF-Inhibitoren als effektive Therapie dieser entzündlichen Systemerkrankungen etabliert (Tabelle 1).
Zu dieser Gruppe gehören die monoklonalen, humanen IgG-Antikörper Adalimumab und Golimumab, der monoklonale, chimäre IgG-Antikörper Infliximab, das humanisierte Fab-Fragment Certolizumab pegol sowie das humane Fusionsprotein Etanercept (Abbildung 2). Alle fünf TNF‐Inhibitoren zielen auf TNF, aber wichtige strukturelle Unterschiede zwischen diesen Wirkstoffen können ihre Wirkungsweise, Pharmakokinetik und Immunogenität beeinflussen [2]. TNF-Inhibitoren werden in vielen Indikationen als First-Line-Biologika empfohlen, so auch bei RA-Patienten, wenn das Ansprechen auf konventionelle krank- heitsmodifizierende Medikamente (DMT / „disease-modifying therapy“) unzureichend ist, eine hohe Krankheitsaktivität oder ungünstige Prognosefaktoren vorliegen [3]. Bei Patienten mit mittelschwerer bis schwerer Psoriasis kann direkt mit TNF-Inhibitoren begonnen werden, wenn konventionelle Therapien keinen ausreichenden Therapieerfolg erwarten lassen [4]. Bei MC-Patienten mit ausgedehntem Dünndarmbefall und/ oder Befall des oberen Gastrointestinaltraktes empfehlen die Leitlinien der Deutschen Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten (DGVS) eine frühzeitige Behandlung mit TNF-Inhibitoren zu erwägen. Bei steroidrefraktärem MC mit mittlerer bis hoher Krankheitsaktivität sollte primär mit TNF-Inhibitoren behandelt werden. Darüber hinaus werden TNF-Inhibitoren beim steroidabhängigen MC als First-Line-Biologika empfohlen [5]. Auch bei CU-Patienten mit mittelschwerer bis schwerer Krankheitsaktivität, die unzureichend auf die Behandlung mit systemischen Steroiden ansprechen bzw. bei denen Kontraindikationen oder Intoleranzen vorliegen, bei Patienten mit einer steroidabhängigen CU sowie bei mittelschwerer bis schwerer CU, die nicht ausreichend auf eine Therapie mit Thiopurinen ansprechen, empfiehlt die DGVS die Behandlung mit TNF-Inhibitoren [6].
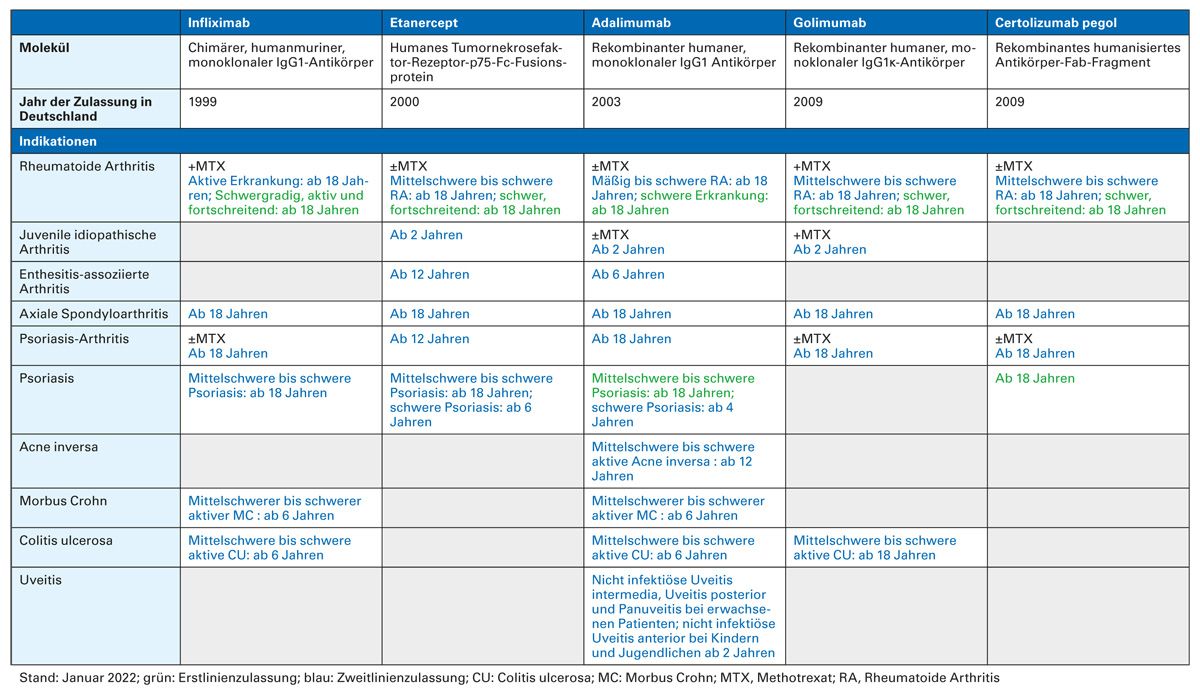
Tab. 1: TNF-Inhibitoren und ihre Indikationen
Kontraindikationen für den Einsatz von TNF-Inhibitoren sind chronische Virusinfektionen, opportunistische Infektionen und schwere Herzinsuffizienz. TNF-Inhibitoren können zu einer demyelinisierenden Neuropathie, Zytopenien, interstitiellen Lungenerkrankungen und vermehrter Infektneigung führen. Zudem zeigt sich bei jahrelanger Therapie ein leicht erhöhtes Risiko für das Auftreten von Malignomen [7-13].
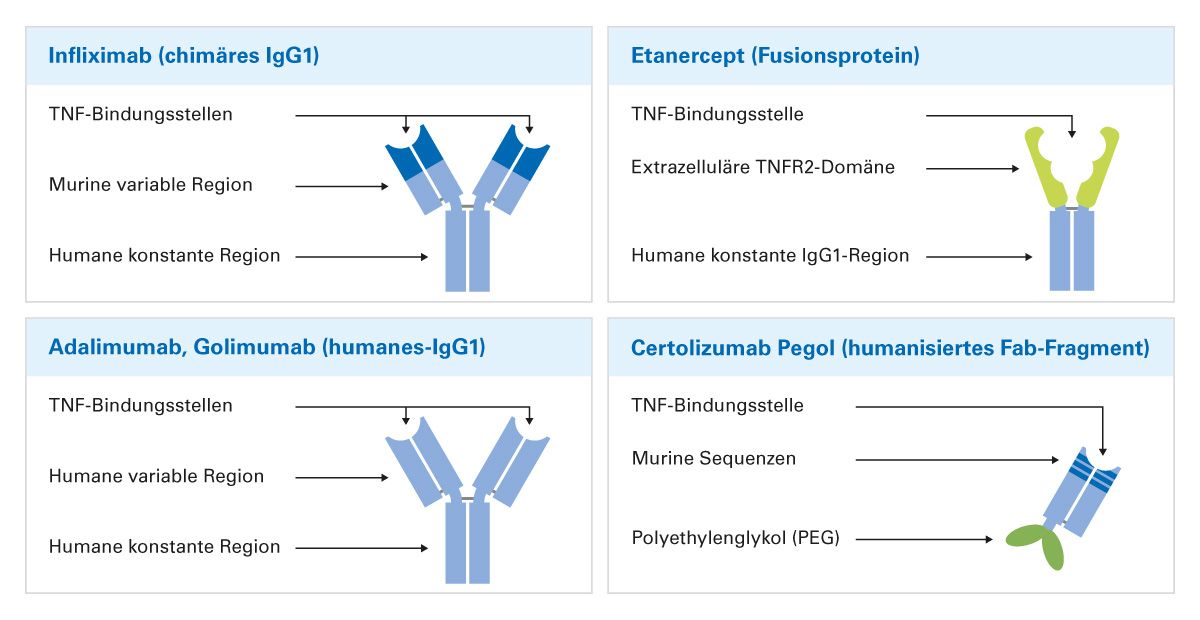
Abb. 2: TNF-Inhibitoren weisen unterschiedliche Strukturen auf
Mono- vs. Kombinationstherapie von TNF- Inhibitoren mit Immunmodulatoren
Bei RA, Psoriasis-Arthritis und juveniler idiopathischer Arthritis können TNF-Inhibitoren als Monotherapie oder in Kombination mit Methotrexat (MTX) eingesetzt werden, sofern keine Unverträglichkeit gegenüber MTX besteht [9-13]. Studienergebnisse zeigten, dass die Kombinationstherapie von Adalimumab und MTX der jeweiligen Monotherapie in allen Endpunkten bei Patienten mit RA überlegen ist (Ansprechrate des American College of Rheumatology mit einer Verbesserung von 50 % [ACR50] unter der Kombinationstherapie: 62 % der Patienten vs. 46 % unter MTX- und 41 % unter Adalimumab-Mono- therapie) [14]. Ebenso war das Gesamtansprechen in einer pädiatrischen klinischen Stu- die mit 171 Kindern mit aktiver polyartikulärer juveniler idiopathischer Arthritis bei der Kombinationstherapie von Adalimumab und MTX allgemein besser als mit Adalimumab allein (94 % vs. 74 %) [15]. Dies kann durch höhere Talspiegel von Adalimumab begründet sein, die in Gegenwart von MTX gemessen wurden [16]. Darüber hinaus kann die Kombination von TNF-Inhibitoren mit MTX das Risiko der Bildung von anti-Drug-Antikörpern (ADAs) verringern [17, 18]. Zur Behandlung von MC war eine Kombinationstherapie mit Adalimumab und Immunmodulatoren einer Monotherapie mit Adalimumab hinsichtlich Wirksamkeit nicht überlegen [19, 20]. Die Kombinationstherapie war jedoch mit einer geringeren Immunogenität assoziiert [19].
Strategien bei Therapieversagen unter TNF-Inhibitor-Therapie
Immunogenität und die Bildung von ADAs werden als Hauptgründe für ein sekundäres Therapieversagen diskutiert, d. h. die Wirksamkeit kann mit fortschreitender Therapie abnehmen. ADAs können die Clearance von zirkulierenden Biologika neutralisieren und/ oder fördern. Dies kann zu niedrigen Medikamentenspiegeln im Serum, Verlust des klinischen Ansprechens, nachlassender Therapietreue und unerwünschten Ereignissen führen [21]. Das Risiko für die Bildung von ADAs ist infolge struktureller Unterschiede (Abbildung 2) zwischen den einzelnen Wirkstoffen unterschiedlich hoch (Anteil Patienten mit ADA-Bildung in den ersten 6 Monaten nach Therapiebeginn: 53 % unter Infliximab-Therapie vs. 19 % unter Adalimumab-Therapie).
Als Fusionsprotein exprimiert Etanercept keine Idiotypen und ist daher der am wenigsten immunogene TNF-Inhibitor (0-7 % der Patienten entwickeln transiente, nicht-neutralisierende ADAs) [21]. Im Fall des Nicht-Ansprechens kann daher bei Patienten mit niedrigen Medikamentenspiegeln aufgrund der ADA-vermittelten Clearance der Wechsel zu einem alternativen TNF‐Inhibitor vorteilhaft sein [22]. Sowohl ein Wechsel innerhalb des Wirkmechanismus (Cycling) als auch zwischen Wirkmechanismen (Swapping) werden durch randomisierte klinische Studien und Daten aus der klinischen Routine gestützt. Zum Beispiel haben Daten der randomisierten EXXELERATE-Studie bei Patienten mit RA sowohl den klinischen Nutzen als auch das gute Sicherheitsprofil einer Umstellung auf einen zweiten TNF-Inhibitor ohne Auswaschphase nach einem primären Versagen eines ersten TNF-Inhibitors belegt. Patienten mit primärem Therapieversagen unter Adalimumab profitierten langfristig von einem Wechsel auf Certolizumab pegol und umgekehrt (Abbildung 3) [23].
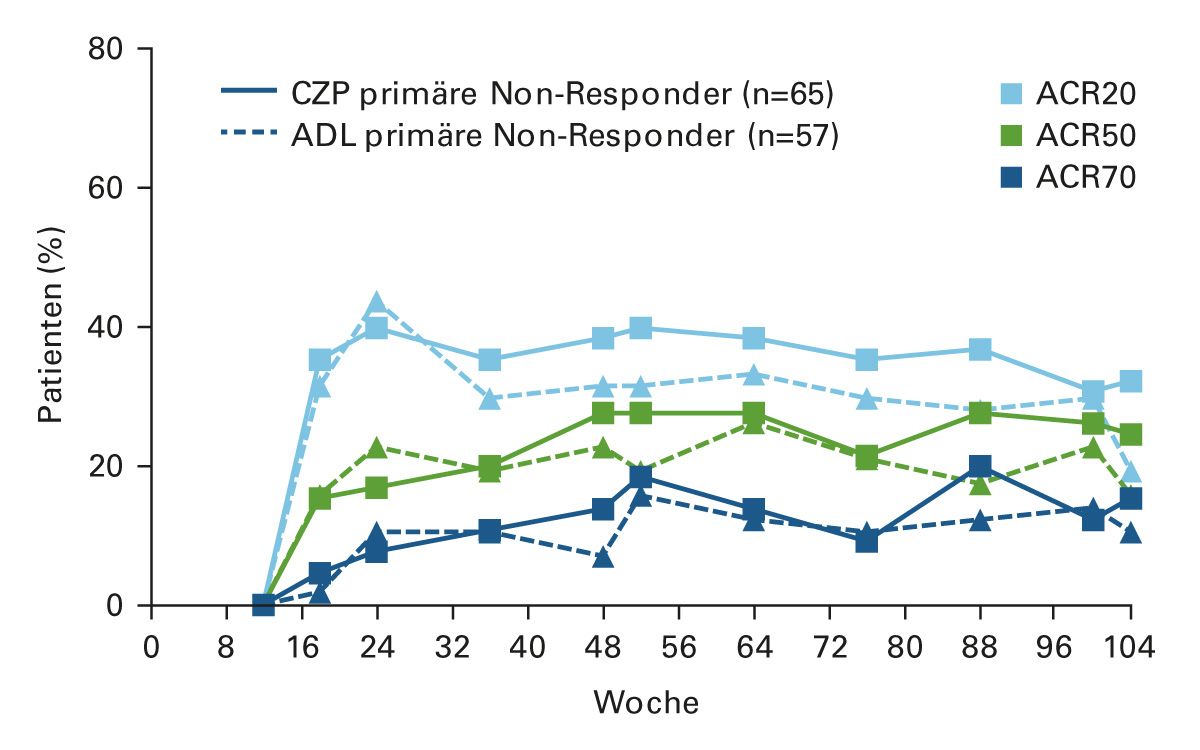
Abbildung 3: Patienten mit primärem Therapieversagen unter Adalimumab oder Certolizumab pegol haben nach dem Wechsel auf den jeweils anderen TNF-Inhibitor ein gutes und langanhaltendes Ansprechen (modifiziert nach [23])
ACR20/50/70, Ansprechraten des American College of Rheumatology mit einer Verbesserung von 20 %/ 50 %/ 70 %; ADL, Adalimumab; CZP, Certolizumab pegol
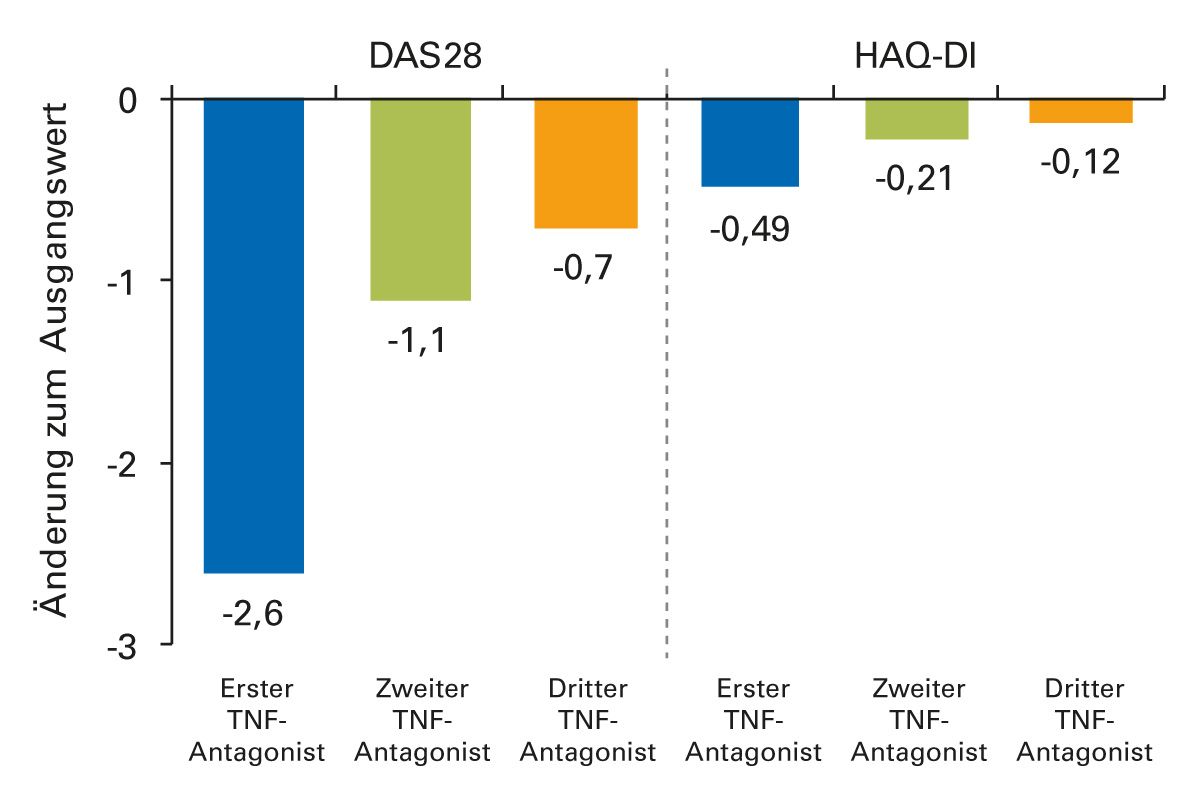
Abbildung 4: Klinisches Ansprechen nach Cycling zwischen TNF-Inhibitoren [26] DAS28, Disease Activity Score 28; HAQ-DI, Health Assessment Questionnaire Disability Index; TNF, Tumornekrosefaktor
Auch ein Therapiewechsel von Etanercept oder Infliximab auf Adalimumab brachte Patienten mit RA einen erheblichen klinischen Nutzen. Zwölf Wochen nach der Umstellung erreichten 60 % von 899 Patienten ein ACR20-Ansprechen und 33 % ein ACR50-Ansprechen [24]. Daten aus dem Biologika-Register der Britischen Rheumatologie-Gesellschaft (BSRBR) zeigten, dass ein signifikanter Anteil Patienten (36 %) beim Cycling auf einen zweiten TNF-Inhibitor die als minimale klinisch differenzierbar definierte Verbesserung von 0,22 Einheiten im Health Assessment Questionnaire (HAQ)-Score erreichte. Dabei profitierten insbesondere Patienten, die frühzeitig innerhalb von 90 Tagen wechselten (46 %) [25]. Das klinische Ansprechen nimmt jedoch mit steigender Zahl von TNF-Inhibitor-Therapien ab, wie Daten einer spanischen Beobachtungsstudie zeigten (Abbildung 4) [26]. Entscheidungen zum Vorgehen bei Therapieversagen werden im Allgemeinen em- pirisch und im Ermessen des Arztes getroffen [27].
Biosimilars – Äquivalente Wirksamkeit und Sicherheitsprofil zum Referenzprodukt
Zur Deckung von Forschungs- und Entwicklungskosten eines biologischen Arzneimittels folgt auf die Zulassung eine Phase des Patentschutzes. Nach Ablauf dieses Zeitraums, in der Regel nach 10-15 Jahren, ist der Markt offen für Biosimilars. Die Europäische Arzneimittelagentur (EMA) definiert ein Biosimilar als ein biologisches Arzneimittel, das als arzneilich wirksamen Bestandteil eine Version des Wirkstoffs eines im europäischen Wirtschaftsraum bereits zugelassenen biologischen Arzneimittels enthält [28]. Die Definition der amerikanischen Zulassungsbehörde FDA enthält mehr Details und charakterisiert ein Biosimilar als ein Biologikum, das – ungeachtet geringfügiger Unterschiede bei klinisch inaktiven Komponenten – eine hohe Ähnlichkeit zu dem Referenzarzneimittel aufweist, wobei zwischen dem Biologikum und dem Referenzarzneimittel keine klinisch bedeutenden Unterschiede hinsichtlich Sicherheitsprofil, Reinheit und Potenz bestehen [29].
Um die Biosimilarität zum Referenzprodukt zu gewährleisten, durchlaufen Biosimilars im Entwicklungsprozess ein aufwändiges schrittweises Prüfverfahren. Die rechtliche Basis für die Zulassung eines Biosimilars ist der Artikel 10 Abs. 4 der Richtlinie 2001/83/EC und weiterhin die Richtlinie 2003/63/EC, Paragraph 4, Teil II des Anhangs I dieser Richtlinie [28]. Bei der Zulassung von Biosimilars muss die Wirksamkeit des biosimilaren Wirkstoffs nicht neu belegt werden, da dies bereits beim Referenzarzneimittel erfolgt ist. Stattdessen muss der Nachweis der gleichen bzw. vergleichbaren klinischen Wirksamkeit wie beim Referenzarzneimittel erbracht werden. Dazu müssen Phase-I- und Phase-III-Studien durchgeführt werden. Anstelle klinischer Studien zu Wirksamkeit und Sicherheitsprofil steht die Analytik und exakte biochemische Charakterisierung als Basis für den Nachweis der Biosimilarität im Vordergrund (Abbildung 5).
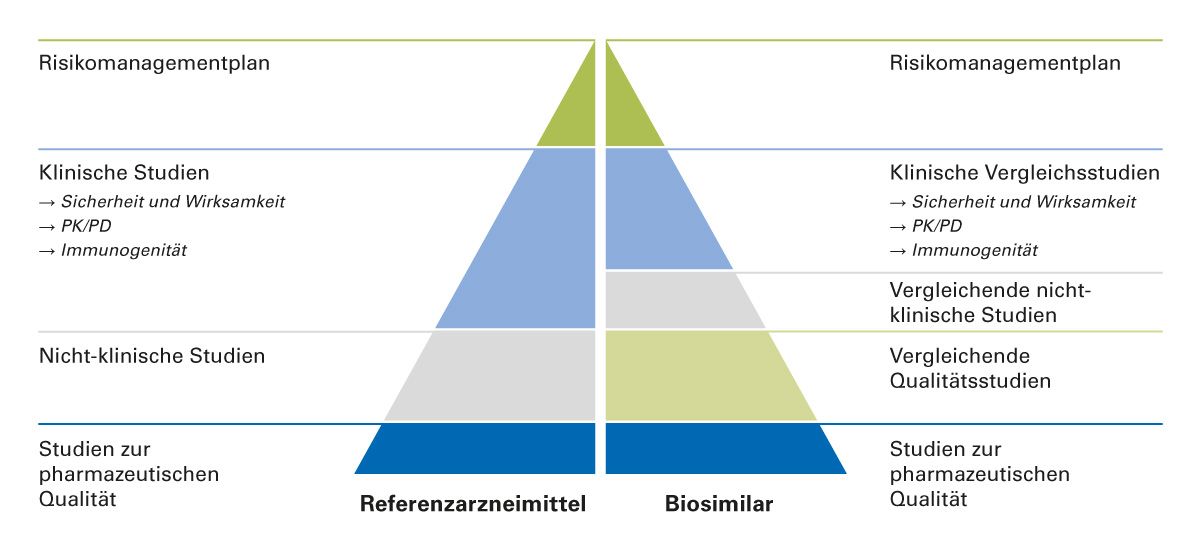
Abb. 5: Vergleich der Datenanforderungen für die Zulassung von Biosimilars gegenüber Referenzarzneimitteln (modifiziert nach [32])
PD, Pharmakodynamik; PK, Pharmakokinetik
Somit sind Studien zur pharmazeutischen Qualität bei den Biosimilars wesentlich aufwändiger als die Studien für ein biologisches Arzneimittel mit neuem Wirkstoff. Vergleichende klinische Studien werden speziell konzipiert, um klinisch relevante Unterschiede in Bezug auf das Sicherheitsprofil und Wirksamkeit zwischen dem Biosimilar und dem Referenzarzneimittel auszuschließen und die Biosimilarität zu bestätigen. Pharmakokinetische Studien sollten in einer homogenen und sensitiven Population durchgeführt werden, um mögliche Unterschiede zwischen dem Biosimilar und dem Referenzarzneimittel festzustellen. Sobald die pharmakokinetische Bioäquivalenz in Phase-I-Studien etabliert wurde, schließt sich eine klinische Phase-III-Studie in einer ausreichend sensitiven Indikation an, d. h. in einer Patientengruppe, bei der mögliche Unterschiede zwischen dem zuzulassenden Biosimilar und dem Referenzarzneimittel am besten beobachtet werden können [30]. Für monoklonale Antikörper wird eine randomisierte doppelblinde Vergleichsstudie mit Parallelgruppendesign gefordert [31].
Mikroheterogenität als Merkmal aller biologischen Arzneimittel
Da biologische Arzneimittel von lebenden Organismen erzeugt werden, die naturbedingt variabel sind, kann der Wirkstoff im endgültigen biologischen Arzneimittel eine geringe Variabilität z. B. im Glykosylierungsmuster aufweisen. Diese Mikroheterogenität muss im festgelegten, akzeptablen Bereich liegen und darf funktionell nicht relevant sein, um dauerhaft die Sicherheit und Wirksamkeit des Arzneimittels zu gewährleisten [32]. Für Biosimilars gilt der gleiche bei Zulassung des Referenzarzneimittels definierte Korridor für Mikroheterogenität wie für eine neue Charge oder eine Änderung im Herstellungsprozess des Referenzarzneimittels. Das heute vom Hersteller des Referenzarzneimittels erhältliche Infliximab ist daher ähnlich, also biosimilar, aber nicht identisch mit dem in den Zulassungsstudien eingesetzten Infliximab. Diese inhärente Variabilität aller Biologika ist für die Wirksamkeit und Sicherheit des Biologikums unkritisch, solange die Veränderungen das bei der Zulassung definierte Ausmaß nicht überschreiten. Hersteller und Zulassungsbehörden beurteilen dies anhand von analytischen Verfahren, die eine höhere Sensitivität als klinische Studien haben, strukturelle und funktionelle Abweichungen aufzudecken [30].
Klinische Vergleichsstudien zu Infliximab, Adalimumab und Etanercept
Für die Zulassung von Infliximab-, Adalimumab- und Etanercept-Biosimilars wurden randomisierte, doppelblinde Phase-III-Parallelgruppenstudien zum Nachweis der vergleichbaren Wirksamkeit und Sicherheitsprofile überwiegend in der Indikation RA durchgeführt.
Nachweis der vergleichenden Wirksamkeit eines Infliximab-Biosimilars
RA-Patienten unter Therapie mit Biosimilar SB2 bzw. Referenz-Infliximab wiesen in Woche 30 äquivalente ACR20-Ansprechraten auf (64,1 % vs. 66,0 %). Die sekundären Wirksamkeitsendpunkte (Disease Activity Score [DAS] 28, ACR50, ACR70) bestätigten die Vergleichbarkeit. Über den gesamten Studienzeitraum von 78 Wochen waren die Sicherheitsprofile und die Immunogenität von Biosimilar und Referenzpräparat vergleichbar [33]. Der Verlauf der Krankheitsaktivität blieb auch bei Umstellung vom Referenzpräparat auf das Biosimilar in Woche 54 in allen drei Gruppen vergleichbar (Abbildung 6); ebenso die Inzidenz unerwünschter Ereignisse bis Woche 78 (36,2 % bei Patienten, die vom Referenz-Infliximab auf SB2 wechselten; 35,6 % bei Patienten, die weiter das Referenzpräparat erhielten; 40,3 % bei Patienten, die weiter das Infliximab-Biosimilar erhielten). Von den Patienten, die bis Woche 54 keine ADAs hatten, entwickelten sich nach der Umstellung auf das Biosimilar ADAs bei 14,6 %. Der Anteil von Patienten, die ADAs entwickelten, war zwischen den Gruppen, die durchgängig das Referenzpräparat bzw. das Biosimilar erhielten, vergleichbar (14,9 vs. 14,1 %) [34].
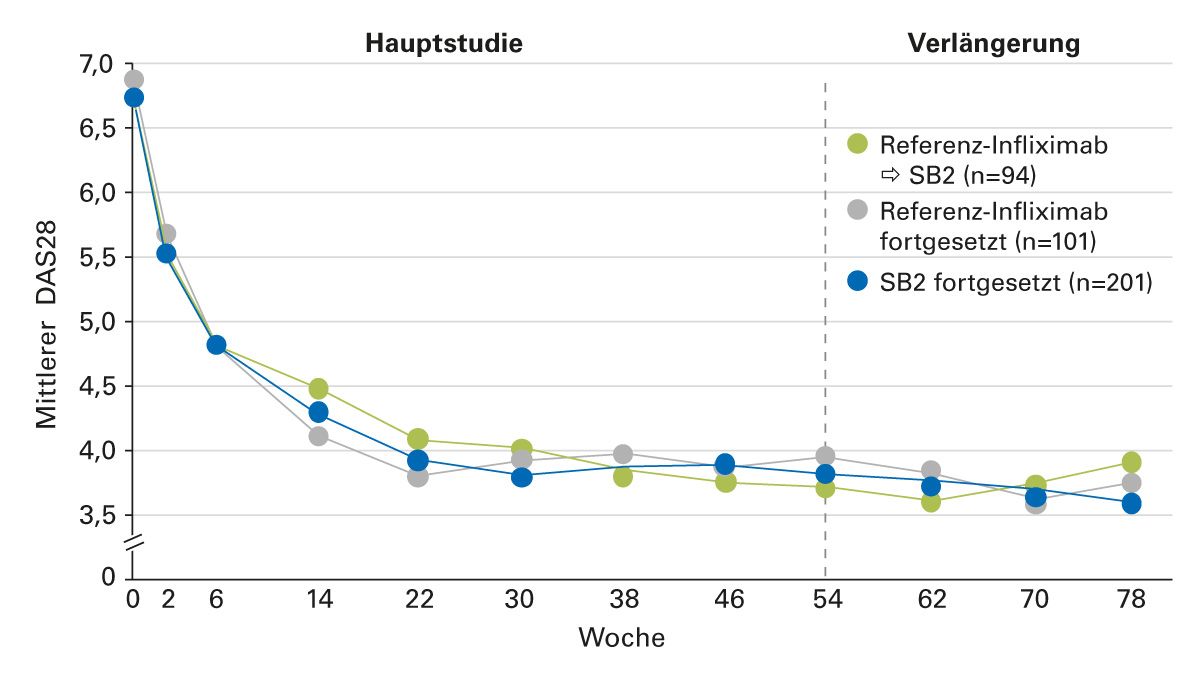
Abb. 6: Verlauf der Krankheitsaktivität bei RA-Patienten im Vergleich unter Therapie mit Referenz- Infliximab, Infliximab-Biosimilar SB2 sowie Umstellung von Referenz-Infliximab auf Infliximab-Biosimilar SB2 nach Woche 54 (Modifiziert nach [34])
DAS28, Disease Activity Score 28
Nachweis der vergleichenden Wirksamkeit eines Adalimumab-Biosimilars
In einer klinischen Phase-III-Studie zur Bewertung von Adalimumab-Biosimilar SB5 und Referenz-Adalimumab wiesen Patienten mit mäßiger bis schwerer RA nach 24 Wochen vergleichbare Ergebnisse zu den Ansprechraten ACR20, ACR50 und ACR70 sowie Pharmakokinetik, Sicherheitsprofil und Immunogenität auf [35]. Diese Ergebnisse konnten auch in der Verlängerung über ein Jahr bestätigt werden (Abbildung 7) [36].
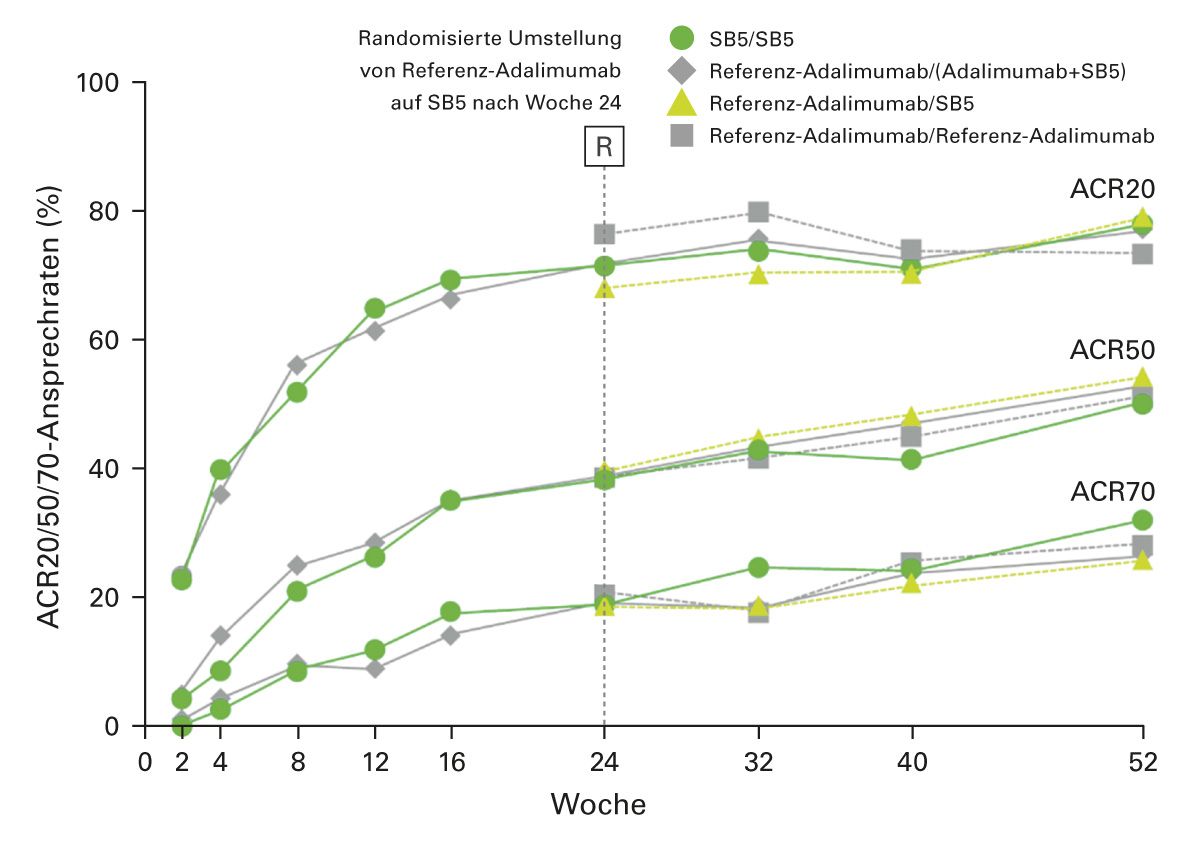
Abb. 7: ACR-Ansprechraten bei RA-Patienten unter Therapie mit Referenz-Adalimumab vs. Adali- mumab-Biosimilar SB5 und nach Umstellung vom Referenz-Adalimumab auf das Biosimilar SB5 (ab Woche 24) (modifiziert nach [36]).
ACR20/50/70, Ansprechraten des American College of Rheumatology mit einer Verbesserung von 20 %/ 50 %/ 70 %
Nachweis der vergleichenden Wirksamkeit eines Etanercept-Biosimilars
Auch für Etanercept bestätigen klinische Daten in der Indikation RA die langfristige Wirksamkeit bei Neueinstellung und Switch vom Referenzprodukt auf ein Biosimilar. Die mittleren DAS28-Werte blieben bei RA-Patienten unter Therapie mit dem Etanercept- Biosimilar SB4 über 100 Wochen stabil, sowohl bei durchgehender Therapie mit dem Etanercept-Biosimilar als auch über 52 Wochen nach Switch vom Referenz-Etanercept auf das Biosimilar (Abbildung 8) [37].
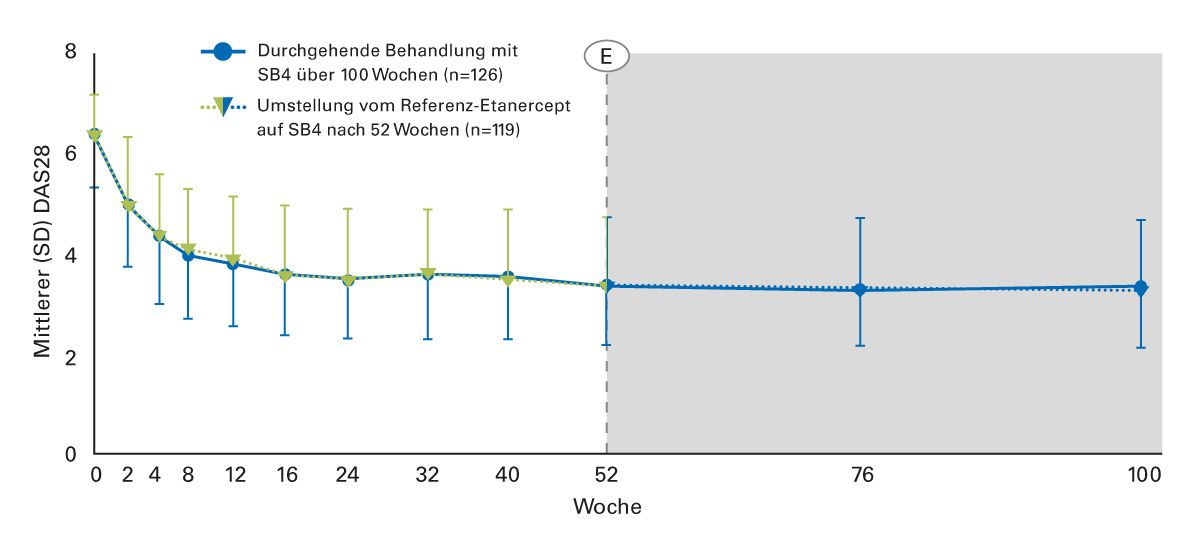
Abb. 8: Verlauf des Disease Activity Score 28 bei RA-Patienten unter durchgehender Therapie mit dem Etanercept-Biosimilar SB4 und nach Umstellung vom Referenz-Etanercept auf das Etanercept-Biosimilar SB4 (modifiziert nach [37]).
DAS28, Disease Activity Score 28; E, Verlängerung; SD, Standardabweichung
Extrapolation der Zulassung auf andere Indikationen
Wenn das Biosimilar den Nachweis der Qualität und Wirksamkeit in einer sensitiven Indikation über Phase-I- und III-Studien erbracht hat und es aus wissenschaftlicher Sicht keine Einwände gibt, kann die Zulassung ohne zusätzliche Phase-III-Studien in anderen Indikationen auf die weiteren Indikationen des Referenzprodukts extrapoliert werden. Dabei muss eine Extrapolation durch die Gesamtheit der in Vergleichbarkeitsstudien generierten wissenschaftlichen Evidenz gestützt werden [32]. Insbesondere müssen der relevante Wirkmechanismus bzw. die an den extrapolierten Indikationen beteiligten Rezeptoren die gleichen sein. Zudem muss das Sicherheitsprofil des Biosimilars ausreichend charakterisiert und eine erhöhte Immunogenität ausgeschlossen sein [38]. Grundsätzlich hat sich die Extrapolation in der Entwicklung von Biologika und in anderen zulassungsrechtlichen Zusammenhängen als allgemein akzeptiertes Konzept etabliert [32].
Biosimilars bringen ökonomische Vorteile für das Gesundheitssystem
Infolge des spezifisch angepassten Entwicklungsprogramms, das auf wissenschaftlichen Erkenntnissen mit dem Referenzarzneimittel beruht, werden Biosimilars in der Regel zu niedrigeren Preisen als das Referenzarzneimittel in den Markt eingeführt. Gleichzeitig sorgte die Einführung von Biosimilars in den europäischen Markt zu wettbewerblichen Preissenkungen nicht nur bei Biosimilars, sondern auch bei den Referenzarzneimitteln [31, 39]. Dabei kann der Markteintritt eines Biosimilars nicht nur den Preis des Referenzarzneimittels beeinflussen, sondern auch das Preisniveau in der gesamten anatomisch-therapeutisch-chemischen Klassifikationsgruppe (ATC) [30]. Seit Januar 2021 ist die vom Gemeinsamen Bundesausschuss beschlossene Festbetragsgruppe „TNFα-Inhibitorengruppe 1 (Stufe 2)“ in Kraft, welche Adalimumab, Etanercept, Golimumab und Certolizumab pegol umfasst [40]. Für Infliximab besteht seit 2017 eine Festbetragsgruppe der Stufe 1 [41]. Der Festbetrag eines Arzneimittels ist der maximale Betrag, den die gesetzlichen Krankenkassen erstatten.
Zum Einsparpotenzial durch Biosimilars wurden viele Rechenmodelle in verschiedenen Versorgungsbereichen publiziert. Besonders hoch ist das Einsparpotenzial bei chronischen Erkrankungen, die eine langfristige Therapie benötigen sowie bei Erkrankungen mit hohen Inzidenzen [42]. Auch TNF-Inhibitoren werden in der Regel langfristig gegen chronisch-entzündliche Erkrankungen eingesetzt. In einem Rechenmodell für den deutschen Markt belaufen sich die jährlichen Einsparungen beispielsweise beim Einsatz von Infliximab-Biosimilars in der Dermatologie, Rheumatologie und Gastroenterologie auf 33,8 Millionen EUR unter der Annahme eines 30 % niedrigeren Preises für das Biosimilar gegenüber dem Referenzprodukt. Damit könnten 2.602 zusätzliche Patienten mit dem Infliximab-Biosimilar behandelt werden [43]. Auf das tatsächliche Einsparpotenzial wirken mehrere Faktoren ein. Neben der Anzahl der in einer Festbetragsgruppe zusammengefassten biologischen Arzneimittel wirkt sich auch die Zeit nach Patentablauf darauf aus. Beispielsweise wurden für Infliximab, zu dem seit mehr als fünf Jahren zum aktuellen Zeitpunkt 4 Biosimilars auf dem Markt sind, mögliche Einsparungen von 3 % berechnet.
Dahingegen beträgt das mögliche Einsparpotenzial für Adalimumab 37 %. Bei Adalimumab ist das allgemeine Preisniveau bereits drei Monate nach Markteintritt des ersten Biosimilars im September 2018 gesunken, zum aktuellen Zeitpunkt sind 6 Biosimilars auf dem deutschen Markt verfügbar [44]. Durch die Festpreisregelung ist zu erwarten, dass die Preisunterschiede zwischen Referenzpräparat und Biosimilar zwar abnehmen, aber in geringerem Maße bestehen bleiben, so dass Biosimilars weiterhin die wirtschaftlich günstigere Variante wären. Sobald kostengünstigere Biosimilar-Präparate zur Verfügung stehen, werden von den kassenärztlichen Vereinigungen Biosimilar-Verordnungsquoten vereinbart, die die ärztliche Verordnung von Biosimilars steuern sollen. Da infolge des Gesetzes für mehr Sicherheit in der Arzneimittelversorgung (GSAV) bei Neueinstellungen von Patienten grundsätzlich ein wirtschaftliches Biosimilar zu verordnen ist, werden die Biosimilar-Verordnungsquoten jährlich überprüft und gegebenenfalls angepasst. Daher kann es infolge der regional unterschiedlichen Quoten zu regionalen Unterschieden in der Marktdurchdringung von Biosimilars im GKV-Markt kommen.
Infolge der Einsparungen können Budgets umgeschichtet werden, um eine optimale Patientenversorgung im Sinne der personalisierten Medizin zu ermöglichen.
Durch den maßgeblichen Beitrag zur Preissenkung ermöglicht die Einführung von Biosimilars eine Entlastung des Gesundheitssystems. Infolge der Einsparungen können Budgets umgeschichtet werden, um eine optimale Patientenversorgung im Sinne der personalisierten Medizin zu ermöglichen. Dies umfasst den Therapiezugang in unterversorgten Bereichen, insbesondere leitliniengerechte Therapien. Zum Beispiel profitieren Patienten in vielen Indikationen von einem frühzeitigen Therapiebeginn, der jedoch aufgrund des Fachärztemangels und damit verbundener langer Wartezeiten auf einen Behandlungstermin häufig nicht realisierbar ist [45, 46].
Voraussetzung für die Realisierung der beschriebenen Einsparpotenziale ist die Akzeptanz von Biosimilars durch die verordnenden Ärzte und die Patienten. Die verfügbaren Biosimilars der TNF-Inhibitoren werden mittlerweile gut in der Versorgung akzeptiert. Beispielsweise lag in der Rheumatologie der Versorgungsanteil von Adalimumab-Biosimilars im Juni 2021 bei 72,5 % und von Etanercept-Biosimilars bei 80,4 %. Infliximab-Biosimilars in der Indikation Morbus Crohn erreichten 87,1 % [47]. Entscheidend für die Akzeptanz bei den Patienten ist das Arzt-Patienten-Gespräch. Dabei sollten die Ärzte auf eine positive Haltung sowie Formulierungsweise gegenüber Biosimilars („Framing“) achten [30].
Wirksamkeit und Sicherheitsprofil von Biosimilars
Gut dokumentierte Wirksamkeit und Sicherheitsprofile der Biosimilars sind essenziell für die Akzeptanz bei den verordnenden Ärzten und den Patienten. Die Hersteller sind entsprechend der EU-Verordnung verpflichtet, einen Risikomanagementplan vorzulegen, der ein Pharmakovigilanz-System umfasst [48]. Seit dem 1. Januar 2011 unterliegen biologische Arzneimittel in den ersten Jahren nach ihrer Zulassung der sogenannten „zusätzlichen Überwachung“. Dieser Status ist durch ein schwarzes auf der Spitze stehendes Dreieck in Fachinformation und Packungsbeilage gekennzeichnet mit dem Pflichttext:
„Dieses Arzneimittel unterliegt einer zusätzlichen Überwachung. Dies ermöglicht eine schnelle Identifizierung neuer Erkenntnisse über die Sicherheit. Angehörige von Gesundheitsberufen sind aufgefordert, jeden Verdachtsfall einer Nebenwirkung zu melden.“ Bei Biosimilars gilt dies für jede mutmaßliche Nebenwirkung, auch wenn diese bereits in der Zusammenfassung der Merkmale des Referenzarzneimittels aufgeführt ist [32].
Real-World-Daten – Erfahrungen aus der klinischen Praxis
Real-World-Daten tragen dazu bei, die in klinischen Studien gewonnenen Erkenntnisse über Wirksamkeit und Sicherheitsprofil eines Arzneimittels im klinischen Praxisalltag zu festigen bzw. neue, bislang unbekannte unerwünschte Arzneimittelwirkungen aufzudecken. Insbesondere bei extrapolierten Indikationen kann Evidenz aus der klinischen Praxis vertrauensbildend sein. In einer Beobachtungsstudie konnten bei CED-Patienten nach dem Wechsel vom Referenz-Infliximab auf das Infliximab-Biosimilar SB2 hohe Remissionsraten über 80 Wochen aufrecht erhalten werden (Abbildung 9). Die Umstellung war gut verträglich und nicht mit immunogenen Reaktionen assoziiert [49].
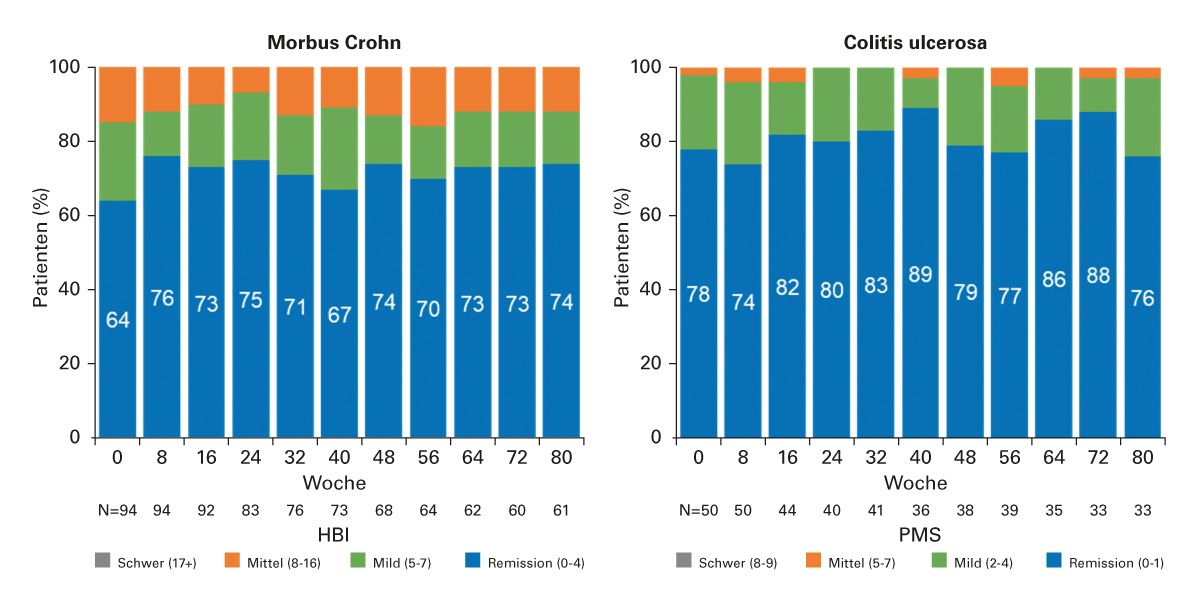
Abb. 9: Klinische Krankheitsindizes im Verlauf nach Umstellung vom Infliximab-Referenzprodukt auf das Infliximab-Biosimilar SB2. (C) HBI bei Morbus Crohn und (D) pMS bei Colitis ulcerosa. Die blaue Spalte zeigt Patienten in klinischer Remission, definiert als HBI ≤4 oder pMS ≤1.
HBI, Harvey-Bradshaw-Index; PMS, partieller Mayo-Score.
Eine weitere Beobachtungsstudie untersuchte die Umstellung vom Referenz-Adalimumab auf das Adalimumab-Biosimilar SB5 bei CED-Patienten. Im Beobachtungszeitraum von einem Jahr wurden keine Unterschiede hinsichtlich Remission, C-reaktivem Protein, fäkalem Calprotectin und Adalimumab-Talspiegeln gefunden. Nach einem Jahr betrug die Retentionsrate noch 83,1 % [50].
Bei der Generierung von Real-World-Daten spielen Register eine wesentliche Rolle. Im deutschen Register RABBIT (Rheumatoide Arthritis – Beobachtung der Biologikathe- rapie) werden Daten zur langfristigen Wirksamkeit und Sicherheitsprofilen von Biologi- katherapien bei Erwachsenen mit RA dokumentiert. Patienten können vom behandelnden Rheumatologen in RABBIT seit 2001 eingeschlossen werden, wenn sie eine Therapie mit einem der zugelassenen Indexpräparate (Referenz-Biologikum, Biosimilar oder Januskinase [JAK]-Inhibitor) oder eine Therapie mit einem synthetischen krankheitsmodifizierenden Antirheumatikum (DMARD) nach Versagen mindestens eines anderen synthetischen DMARDs (Kontrollgruppe) beginnen. Die geplante Beobachtungsdauer beträgt bis zu 10 Jahre, unabhängig von weiteren Therapiewechseln [51]. Von 1.751 Patienten mit einer mindestens 6-monatigen Therapiedauer mit dem Referenz-Etanercept wurden im Beobachtungsverlauf 113 Patienten auf Etanercept-Biosimilars umgestellt. Als häufigsten Grund für die Umstellung wurden Therapiekosten angegeben (79 %). Die Retentionsrate unterschied sich nicht signifikant zwischen Patienten mit und ohne The- rapiewechsel (23 % vs. 17 % mit Therapieabbruch nach 1 Jahr) [52].
Parallel zum RABBIT-Register wurde 2017 für Patienten mit axialer Spondyloarthritis oder Psoriasis-Arthritis das Web-basierte RABBIT-SpA-Register initiiert. Auch hier zeigt sich im Verlauf eine zunehmende Verordnung von Biosimilars [51].
In Dänemark besteht seit 2016 die Verpflichtung, bei Neueinstellung oder Therapie- wechsel auf ein Biologikum ein Biosimilar einzusetzen. Das dänische Register DANBIO zeigte bei Patienten mit RA keine klinisch relevante Zunahme der Inanspruchnahme ambulanter Gesundheitsressourcen innerhalb von 6 Monaten nach Umstellung auf ein Infliximab-Biosimilar [53]. Zudem wurden beim Vergleich umgestellter Patienten mit einer historischen Kontrollkohorte keine Auswirkungen der Umstellung auf die Krankheitsaktivität und die Häufigkeit akuter Erkrankungsschübe registriert [54].
Mehrfacher Switch – kein Einfluss auf Wirksamkeit, Sicherheitsprofil und Immunogenität
Während per Definition Biosimilarität der physikalisch-chemischen und funktionellen Eigenschaften des Biosimilars mit dem Referenzprodukt besteht, kann es Unterschiede in Bezug auf sonstige Bestandteile, Lagerung und Haltbarkeit geben, wie Tabelle 2 am Beispiel Adalimumab zeigt. Mehrfache Umstellungen zwischen Biosimilar und Referenzprodukt wurden in den Indikationen Psoriasis und CED untersucht. Bis zu viermalige Wechsel zwischen Referenz-Adalimumab und Adalimumab-Biosimilar GP2017 ergaben keine nachweisbaren Auswirkungen auf Wirksamkeit, Sicherheitsprofil oder Immunogenität von Psoriasis-Patienten über einen Zeitraum von 51 Wochen [55]. Bei Patienten mit mittelschwerer bis schwerer chronischer Plaque-Psoriasis zeigten sich nach insgesamt 52 Wochen und dreifachem Switch zwischen Referenz-Etanercept und dem Etanercept-Biosimilar GP2015 keine Unterschiede bezüglich Wirksamkeit, Sicherheitsprofil und Immunogenität zwischen den durchgängig behandelten und den mehrfach umgestellten Patienten [56, 57].
Weder der Wechsel vom Referenz-Adalimumab auf das Adalimumab-Biosimilar ABP501 als auch der erneute Wechsel auf ein weiteres Adalimumab-Biosimilar SB5 führte bei Patienten mit Morbus Crohn zu einem Wirkverlust [58]. Umstellungen vom Referenz-Infliximab auf das Infliximab-Biosimilar SB2 sowie mehrfache Wechsel zwischen Referenz-Infliximab und zwei verschiedenen Biosimilars wiesen keine Unterschiede hinsichtlich Wirksamkeit bei CED-Patienten auf [59]. Bei der mehrfachen Umstellung zwischen unterschiedlichen Infliximab-Biosimilars über 3 Jahre im Routine-Alltag bei Patienten mit CED hatte die Anzahl verschiedener Infliximab-Biosimilars keinen Einfluss auf die Inzidenz von ADAs. Das Risiko eines The- rapieabbruchs war jedoch bei Patienten höher, die ADAs entwickelten [60].
Nocebo-Effekt als Grund für vorzeitige Therapieabbrüche
Im Vergleich zu doppelblinden randomisierten Studien mit einer Therapie-Abbruchrate von <10 % wurden bei Beobachtungsstudien aus dem Praxisalltag bei der Umstellung auf Biosimilars teilweise höhere Raten an vorzeitigen Therapieabbrüchen (15 – 30 %) beobachtet, insbesondere bei Infliximab, Etanercept und Adalimumab. Dies wird auf Nocebo-Effekte zurückgeführt – unerklärbare, negative Effekte bei der Anwendung eines Arzneimittels, die auf der Basis von negativen Erwartungen des Patienten über schädliche Auswirkungen einer Therapie entstehen können [30]. Da sich Nocebo-Effekte negativ auf die Adhärenz, Nebenwirkungen und die Symptomlinderung während der medikamentösen Behandlung auswirken können, ist es essenziell, diese Effekte durch ausführliche Information und Beratung der Patienten zu minimieren bzw. zu vermeiden. Beispielsweise zeigte sich in einer offenen Switch-Studie in den Niederlanden, dass eine intensivierte Informationsstrategie zu niedrigeren Abbruchsraten nach der Umstellung auf Biosimilars von Etanercept oder Infliximab bei Patienten mit rheumatischen Erkran- kungen führt (6 % vs. 24 %) [61]. Dagegen ergab eine retrospektive Datenanalyse, dass ein systematischer Wechsel vom Referenz-Etanercept auf das Etanercept-Biosimilar SB4 bei Patienten mit RA, PsA oder axialer Spondyloarthritis nicht mit Veränderungen der Krankheitsaktivität oder Funktion assoziiert war, unabhängig davon, ob der Patient über die Umstellung informiert wurde oder nicht [62]. Dennoch ist eine gute proaktive Kommunikation zwischen Arzt und Patient als wichtiger Schritt zur Vermittlung von Vertrauen des Patienten in die Biosimilars ratsam [30].
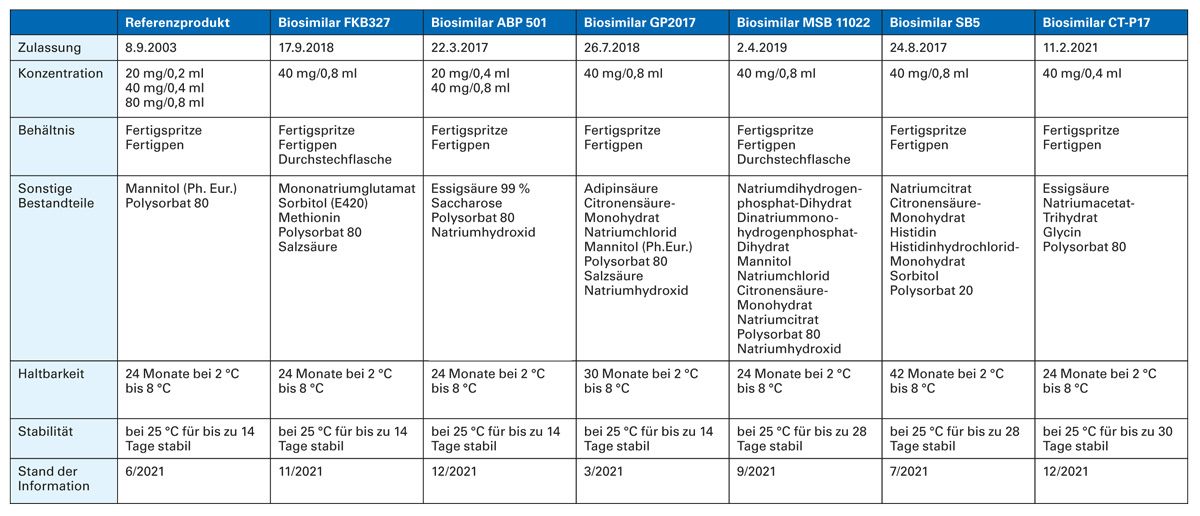
Tab. 2: Adalimumab – Charakteristika des Referenzprodukts und der Biosimilars (Stand Januar 2022) [64]
Automatische Substitution von Biologika
Entscheidungen zur automatischen Substitution von Referenzarzneimitteln durch Biosimilars trifft nicht die EMA, sondern jeder Mitgliedsstaat der EU, da es sich um eine Frage des nationalen Rechts handelt. Anders als in Dänemark und einigen anderen EU-Ländern, ist die automatische Substitution aktuell (Oktober 2021) in Deutschland für Biologika und Biosimilars nicht zulässig, jedoch ab August 2022 geplant. Aktuell darf nur ein Austausch auf ein „Bioidentical“ erfolgen. Dabei handelt es sich um Produkte, die im Rahmenvertrag über die Arzneimittelversorgung nach § 129 Absatz 2 SGB V, Anlage 1s als austauschbar definiert sind [63]. Bioidenticals sind identische Kopien von Biosimilars, die unter verschiedenen Handelsnamen von verschiedenen Firmen vertrieben werden. Sie werden mit identischem Herstellungs- und Formulierungsprozess produziert und sind wirkstoffgleich. Die Ausweitung der Aut-idem-Regelung auf Biologika und Biosimilars wird aktuell kontrovers diskutiert und eine endgültige Entscheidung in 2022 erwartet [30].
Fazit
Biosimilars bringen eine Vielzahl ökonomischer Vorteile für das Gesundheitssystem mit. Damit Einsparpotenziale realisiert werden können, ist es wichtig, dass Biosimilars von den verordnenden Ärzten akzeptiert und diese Akzeptanz dem Patienten im Gespräch weitervermittelt wird. Die Zulassung von Biosimilars erfolgt auf der Grundlage einer umfassenden Vergleichbarkeitsprüfung. Bislang ist in keiner klinischen Studie, die den Switch einer laufenden biologischen Therapie auf ein Biosimilar untersucht hat, ein signifikanter Unterschied hinsichtlich der Wirksamkeit oder Verträglichkeit zwischen Biosimilar und Referenzarzneimittel festgestellt worden.
Literatur
1. Carswell EA, Old LJ, Kassel RL et al. (1975) An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A 72 (9):3666-3670. doi:10.1073/pnas.72.9.3666
2. Armuzzi A, Lionetti P, Blandizzi C et al. (2014) anti-TNF agents as therapeutic choice in immune-mediated inflammatory diseases: focus on adalimumab. Int J Immunopathol Pharmacol 27 (1 Suppl):11-32. doi:10.1177/03946320140270s102
3. Fiehn C, Holle J, Iking-Konert C et al. (2018) S2e-Leitlinie: Therapie der rheumatoiden Arthritis mit krankheitsmodifizie- renden Medikamenten. Zeitschrift für Rheumatologie 77 (2):35-53. doi:10.1007/s00393-018-0481-y
4. Nast A, Altenburg A, Augustin M et al. (2021) Deutsche S3-Leitlinie zur Therapie der Psoriasis vulgaris, adaptiert von EuroGuiDerm – Teil 1: Therapieziele und Therapieempfehlungen. JDDG: Journal der Deutschen Dermatologischen Gesellschaft 19 (6):934-951. doi:https://doi.org/10.1111/ddg.14508_g
5. Sturm A, Atreya R, Bettenworth D et al. Aktualisierte S3-Leitlinie „Diagnostik und Therapie des Morbus Crohn“ der Deutschen Gesellschaft für Gastroenterologie, Verdauungs- und Stoffwechselkrankheiten (DGVS). August 2021 – AWMF-Registernummer: 021-004. https://www.dgvs.de/wp-content/uploads/2021/08/Leitlinie-LL-MC_25.08.2031_ final.pdf. Aufgerufen am: 13.10.2021
6. Kucharzik T, Koletzko S, Kannengießer K, Dignaß A (2020) Colitis ulcerosa – Diagnostische und therapeutische
Algorithmen. Dtsch Arztebl International 117 (33-34):564-573
7. Schneider M, Baseler G, Funken O et al. (2020) [Management of early rheumatoid arthritis : Interdisciplinary guideline]. Z Rheumatol 79 (Suppl 1):1-38. doi:10.1007/s00393-020-00775-6
8. Smolen JS, Landewé R, Breedveld FC et al. (2014) EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease-modifying antirheumatic drugs: 2013 update. Ann Rheum Dis 73 (3):492-
509. doi:10.1136/annrheumdis-2013-204573
9. Golimumab Fachinformation (Stand Oktober 2021). https://www.fachinfo.de/suche/stoff/124940/Golimumab. Aufgerufen am: 10.1.2022
10. Certolizumab pegol Fachinformation (Stand September 2021). https://www.fachinfo.de/suche/stoff/124939/Certoli- zumab%20Pegol. Aufgerufen am: 10.1.2022
11. Adalimumab (Referenz) Fachinformation (Stand Juni 2021). https://www.fachinfo.de/suche/fi/020795. Aufgerufen am: 10.1.2022
12. Etanercept (Referenz) Fachinformation (Stand Mai 2021). https://www.fachinfo.de/suche/fi/011927. Aufgerufen am:
10.1.2022
13. Infliximab (Referenz) Fachinformation (Stand Oktober 2021). https://www.fachinfo.de/suche/fi/022911. Aufgerufen am: 10.1.2022
14. Breedveld FC, Weisman MH, Kavanaugh AF et al. (2006) The PREMIER study: A multicenter, randomized, double-blind clinical trial of combination therapy with adalimumab plus methotrexate versus methotrexate alone or adalimumab alone in patients with early, aggressive rheumatoid arthritis who had not had previous methotrexate treatment. Arthritis Rheum 54 (1):26-37. doi:10.1002/art.21519
15. Lovell DJ, Ruperto N, Goodman S et al. (2008) Adalimumab with or without methotrexate in juvenile rheumatoid arthritis. N Engl J Med 359 (8):810-820. doi:10.1056/NEJMoa0706290
16. Pouw MF, Krieckaert CL, Nurmohamed MT et al. (2015) Key findings towards optimising adalimumab treatment: the concentration-effect curve. Ann Rheum Dis 74 (3):513-518. doi:10.1136/annrheumdis-2013-204172
17. Busard C, Zweegers J, Limpens J, Langendam M, Spuls PI (2014) Combined use of systemic agents for psoriasis: a systematic review. JAMA Dermatol 150 (11):1213-1220. doi:10.1001/jamadermatol.2014.1111
18. Pappas DA, Shan Y, Lesperance T et al. (2020) Maintenance of Sustained Low Disease Activity or Remission in Patients With Rheumatoid Arthritis Treated With Etanercept Monotherapy: Results from the Corrona Registry. ACR Open Rheumatol 2 (10):588-594. doi:10.1002/acr2.11168
19. Chalhoub JM, Rimmani HH, Gumaste VV, Sharara AI (2017) Systematic Review and Meta-analysis: Adalimumab Monotherapy Versus Combination Therapy with Immunomodulators for Induction and Maintenance of Remission and Response in Patients with Crohn‘s Disease. Inflamm Bowel Dis 23 (8):1316-1327. doi:10.1097/mib.0000000000001203
20. Matsumoto T, Motoya S, Watanabe K et al. (2016) Adalimumab Monotherapy and a Combination with Azathioprine for
Crohn‘s Disease: A Prospective, Randomized Trial. J Crohns Colitis 10 (11):1259-1266. doi:10.1093/ecco-jcc/jjw152
21. Mehta P, Manson JJ (2020) What Is the Clinical Relevance of TNF Inhibitor Immunogenicity in the Management of
Patients With Rheumatoid Arthritis? Front Immunol 11:589. doi:10.3389/fimmu.2020.00589
22. Bendtzen K (2015) Immunogenicity of Anti-TNF-α Biotherapies: I. Individualized Medicine Based on Immunopharmaco- logical Evidence. Front Immunol 6:152. doi:10.3389/fimmu.2015.00152
23. Smolen JS, Burmester GR, Combe B et al. (2016) Head-to-head comparison of certolizumab pegol versus adalimumab in rheumatoid arthritis: 2-year efficacy and safety results from the randomised EXXELERATE study. Lancet 388 (10061):2763-2774. doi:10.1016/s0140-6736(16)31651-8
24. Bombardieri S, Ruiz AA, Fardellone P et al. (2007) Effectiveness of adalimumab for rheumatoid arthritis in patients with a history of TNF-antagonist therapy in clinical practice. Rheumatology 46 (7):1191-1199. doi:10.1093/rheumatology/ kem091
25. Hyrich KL, Lunt M, Dixon WG, Watson KD, Symmons DP (2008) Effects of switching between anti-TNF therapies on HAQ response in patients who do not respond to their first anti-TNF drug. Rheumatology (Oxford) 47 (7):1000-1005. doi:10.1093/rheumatology/ken127
26. Navarro-Sarabia F, Ruiz-Montesinos D, Hernandez B et al. (2009) DAS-28-based EULAR response and HAQ improvement in rheumatoid arthritis patients switching between TNF antagonists. BMC Musculoskelet Disord 10:91. doi:10.1186/1471-2474-10-91
27. Rubbert-Roth A, Szabó MZ, Kedves M et al. (2019) Failure of anti-TNF treatment in patients with rheumatoid arthritis: The pros and cons of the early use of alternative biological agents. Autoimmun Rev 18 (12):102398. doi:10.1016/j. autrev.2019.102398
28. European Medicines Agency. Guideline on similar biological medicinal products. https://www.ema.europa.eu/en/do- cuments/scientific-guideline/guideline-similar-biological-medicinal-products-rev1_en.pdf. Aufgerufen am: 26.7.2021
29. FDA Biosimilar and Interchangeable Products. US Food & Drug Administration. https://www.fda.gov/drugs/biosimi- lars/biosimilar-and-interchangeable-products. Aufgerufen am: 26.7.2021
30. Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ). Biosimilars. Leitfaden der Arzneimittelkommission der deutschen Ärzteschaft (AkdÄ). 2. Auflage, Version 1.0. Januar 2021. https://www.akdae.de/Arzneimitteltherapie/LF/ PDF/Biosimilars.pdf. Aufgerufen am: 26.7.2021
31. European Medicines Agency. Guideline on similar biological medicinal products containing monoclonal antibodies
– non-clinical and clinical issues (2012). https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-si- milar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical_en.pdf. Aufgerufen am: 26.7.2021
32. European Medicines Agency. Biosimilars in der EU. Leitfaden für medizinische Fachkräfte (2019). https://www.ema. europa.eu/en/documents/leaflet/biosimilars-eu-information-guide-healthcare-professionals_de.pdf. Aufgerufen am: 26.7.2021
33. Choe JY, Prodanovic N, Niebrzydowski J et al. (2017) A randomised, double-blind, phase III study comparing SB2, an infliximab biosimilar, to the infliximab reference product Remicade in patients with moderate to severe rheumatoid arthritis despite methotrexate therapy. Ann Rheum Dis 76 (1):58-64. doi:10.1136/annrheumdis-2015-207764
34. Smolen JS, Choe J-Y, Prodanovic N et al. (2018) Safety, immunogenicity and efficacy after switching from reference infliximab to biosimilar SB2 compared with continuing reference infliximab and SB2 in patients with rheumatoid arthritis: results of a randomised, double-blind, phase III transition study. Annals of the Rheumatic Diseases 77 (2):234. doi:10.1136/annrheumdis-2017-211741
35. Weinblatt ME, Baranauskaite A, Niebrzydowski J et al. (2018) Phase III Randomized Study of SB5, an Adalimumab Biosimilar, Versus Reference Adalimumab in Patients With Moderate-to-Severe Rheumatoid Arthritis. Arthritis Rheumatol 70 (1):40-48. doi:10.1002/art.40336
36. Weinblatt ME, Baranauskaite A, Dokoupilova E et al. (2018) Switching From Reference Adalimumab to SB5 (Adalimumab Biosimilar) in Patients With Rheumatoid Arthritis: Fifty-Two-Week Phase III Randomized Study Results. Arthritis Rheumatol 70 (6):832-840. doi:10.1002/art.40444
37. Emery P, Vencovský J, Sylwestrzak A et al. (2017) Long-term efficacy and safety in patients with rheumatoid arthritis continuing on SB4 or switching from reference etanercept to SB4. Annals of the Rheumatic Diseases 76 (12):1986. doi:10.1136/annrheumdis-2017-211591
38. Weise M, Kurki P, Wolff-Holz E, Bielsky MC, Schneider CK (2014) Biosimilars: the science of extrapolation. Blood 124 (22):3191-3196. doi:10.1182/blood-2014-06-583617
39. IMS The impact of biosimilar competition in Europe (2017). Quintiles IMS. https://www.medicinesforeurope.com/ wp-content/uploads/2017/05/IMS-Biosimilar-2017_V9.pdf. Aufgerufen am: 30.7.2021
40. Gemeinsamer Bundesausschuss. Arzneimittel-Richtlinie/Anlage IX und X: Festbetragsgruppenbildung und Ver- gleichsgrößenaktualisierung – TNF-alpha-Inhibitoren, Gruppe 1, in Stufe 2. https://www.g-ba.de/beschluesse/4550/. Aufgerufen am: 30.7.2021
41. Gemeinsamer Bundesausschuss. Beschluss des Gemeinsamen Bundesausschusses über eine Änderung der Arzneimittel-Richtlinie (AM-RL): Anlage IX – Festbetragsgruppenbildung Infliximab, Gruppe 1, in Stufe 1 nach § 35 Abs. 1 SGB V. https://www.g-ba.de/downloads/39-261-3132/2017-11-17_AM-RL-IX_Infliximab_G1S1_BAnz.pdf. Aufgerufen am: 11.10.2021
42. Hübel K, Kron F, Lux MP (2020) Biosimilars in oncology: Effects on economy and therapeutic innovations. Eur J Cancer 139:10-19. doi:10.1016/j.ejca.2020.07.037
43. Jha A, Upton A, Dunlop WC, Akehurst R (2015) The Budget Impact of Biosimilar Infliximab for the Treatment of Autoimmune Diseases in Five European Countries. Adv Ther 32 (8):742-756. doi:10.1007/s12325-015-0233-1
44. Vogler S, Schneider P, Zuba M, Busse R, Panteli D (2021) Policies to Encourage the Use of Biosimilars in European Countries and Their Potential Impact on Pharmaceutical Expenditure. Front Pharmacol 12:625296. doi:10.3389/ fphar.2021.625296
45. Kyburz D, Gabay C, Michel BA, Finckh A (2011) The long-term impact of early treatment of rheumatoid arthritis on radiographic progression: a population-based cohort study. Rheumatology (Oxford) 50 (6):1106-1110. doi:10.1093/ rheumatology/keq424
46. Augustin M, Glaeske G, Hagenström K Psoriasisreport. Ergebnisse von Routinedaten-Analysen der Techniker Kranken- kasse aus den Jahren 2017–2019. https://www.uke.de/dateien/institute/versorgungsforschung-in-der-dermatologie- und-bei-pflegeberufen-%28ivdp%29/dokumente/psoriasisreport_2021.pdf. Aufgerufen am: 14.10.2021
47. AG Pro Biosimilars. INSIGHT Health GKV-Abrechnungsdaten (NVI-Plus) ambulanter Fertigarzneimittelmarkt (inkl. Zubereitungen). https://probiosimilars.de/img_upload/2021/07/Marktdaten-Biosimilars_Juni-2021.pdf. Aufgerufen am: 2.8.2021
48. Arbeitsgemeinschaft ProBiosmilars. Handbuch Biosimilars (2019). https://probiosimilars.de/img_upload/2021/04/ Handbuch-Biosimilars_Oktober-2019.pdf. Aufgerufen am: 4.8.2021
49. Fischer S, Cohnen S, Klenske E et al. (2021) Long-term effectiveness, safety and immunogenicity of the biosimilar SB2 in inflammatory bowel disease patients after switching from originator infliximab. Therap Adv Gastroenterol 14:1756284820982802. doi:10.1177/1756284820982802
50. Derikx L, Dolby HW, Plevris N et al. (2021) Effectiveness and Safety of Adalimumab Biosimilar SB5 in IBD: Outcomes in Originator to SB5 Switch, Double Biosimilar Switch and Bio-Naieve SB5 Observational Cohorts. J Crohns Colitis. doi:10.1093/ecco-jcc/jjab100
51. Strangfeld A, Regierer A, A Z (2021) Biosimilars in den deutschen Biologika-Registern RABBIT und RABBIT-SpA. In: Kompendium Biosimilars 2021. Thieme, pp 24-33
52. Baganz L, Strangfeld A, Herzer P et al. (2019) SAT0134 Comparing real-world retention rates in a matched cohort of rheumatoid arthritis patients who either remained on the etanercept originator or switched to a biosimilar. Annals of the Rheumatic Diseases 78 (Suppl 2):1136. doi:10.1136/annrheumdis-2019-eular.4026
53. Glintborg B, Sørensen J, Hetland ML (2018) Does a mandatory non-medical switch from originator to biosimilar infli- ximab lead to increased use of outpatient healthcare resources? A register-based study in patients with inflammatory arthritis. RMD Open 4 (2):e000710. doi:10.1136/rmdopen-2018-000710
54. Glintborg B, Loft AG, Omerovic E et al. (2019) To switch or not to switch: results of a nationwide guideline of mandatory switching from originator to biosimilar etanercept. One-year treatment outcomes in 2061 patients with inflammatory arthritis from the DANBIO registry. Annals of the Rheumatic Diseases 78 (2):192. doi:10.1136/ annrheumdis-2018-213474
55. Blauvelt A, Lacour JP, Fowler JF, Jr. et al. (2018) Phase III randomized study of the proposed adalimumab biosimilar GP2017 in psoriasis: impact of multiple switches. Br J Dermatol 179 (3):623-631. doi:10.1111/bjd.16890
56. Gerdes S, Thaçi D, Griffiths CEM et al. (2018) Multiple switches between GP2015, an etanercept biosimilar, with originator product do not impact efficacy, safety and immunogenicity in patients with chronic plaque-type psoriasis: 30-week results from the phase 3, confirmatory EGALITY study. J Eur Acad Dermatol Venereol 32 (3):420-427. doi:10.1111/jdv.14605
57. Griffiths CEM, Thaçi D, Gerdes S et al. (2017) The EGALITY study: a confirmatory, randomized, double-blind study comparing the efficacy, safety and immunogenicity of GP2015, a proposed etanercept biosimilar, vs. the originator product in patients with moderate-to-severe chronic plaque-type psoriasis. Br J Dermatol 176 (4):928-938. doi:10.1111/ bjd.15152
58. Ribaldone DG, Tribocco E, Rosso C et al. (2021) Switching from Biosimilar to Biosimilar Adalimumab, Including Multiple
Switching, in Crohn‘s Disease: A Prospective Study. J Clin Med 10 (15). doi:10.3390/jcm10153387
59. Macaluso FS, Fries W, Viola A et al. (2021) The SPOSIB SB2 Sicilian Cohort: Safety and Effectiveness of Infliximab Biosimilar SB2 in Inflammatory Bowel Diseases, Including Multiple Switches. Inflamm Bowel Dis 27 (2):182-189. doi:10.1093/ibd/izaa036
60. Lauret A, Moltó A, Abitbol V et al. (2020) Effects of successive switches to different biosimilars infliximab on immunogenicity in chronic inflammatory diseases in daily clinical practice. Semin Arthritis Rheum 50 (6):1449-1456. doi:10.1016/j.semarthrit.2020.02.007
61. Tweehuysen L, Huiskes VJB, Bemt BJFvd, Hoogen FHJvd, Broeder AAd (2017) FRI0200 Higher acceptance and persistence rates after biosimilar transitioning in patients with a rheumatic disease after employing an enhanced com- munication strategy. Annals of the Rheumatic Diseases 76 (Suppl 2):557. doi:10.1136/annrheumdis-2017-eular.2889
62. Kiltz U, Pudelko JC, Tsiami S, Baraliakos X, Braun J (2021) Non-medical switching from reference to biosimilar etanercept – no evidence for nocebo effect: a retrospective analysis of real-life data. Clin Exp Rheumatol
63. GKV-Spitzenverband. Rahmenvertrag über die Arzneimittelversorgung nach § 129 Absatz 2 SGB V in der Fassung vom
01. April 2020. https://www.gkv-spitzenverband.de/media/dokumente/krankenversicherung_1/arzneimittel/rahmen- vertraege/apotheken/Rahmenvertrag_nach_129_Abs.2_SGB_V_vom_01.04.2020_.pdf. Aufgerufen am: 14.10.2021
64. Adalimumab Fachinformation. https://www.fachinfo.de/suche/stoff/124643/Adalimumab. Aufgerufen am: 11.1.2022
Bildquellen
Titel: © ustas – stock.adobe.com
Tutorielle Unterstützung
Die tutorielle Unterstützung der Fortbildungsteilnehmer erfolgt durch unseren ärztlichen Leiter Dr. med. Alexander Voigt in Zusammenarbeit mit der arztCME-Redaktion. Inhaltliche Fragen können über das Kommentarfeld, direkt per Mail an service@arztcme.de oder via Telefon unter Tel.: +49(0)180-3000759 gestellt werden. Inhaltliche Fragen werden von unserem ärztlichen Leiter bzw. nach Rücksprache mit diesem und evtl. dem Autor auch von der arztCME-Redaktion beantwortet.
Technischer Support
Der technische Support der arztCME-Online-Akademie erfolgt durch geschulte Mitarbeiterinnen und Mitarbeiter des Betreibers health&media GmbH unter der E-Mail-Adresse technik@arztcme.de oder via Telefon unter Tel.: 49(0)180-3000759.