Friedreich-Ataxie – Aktuelles zu Ursache, Diagnostik und neuen Therapieoptionen
Interessengebiete: Allgemeinmedizin und Innere Medizin, Kinder- und Jugendmedizin, Neurologie
Die Friedreich-Ataxie ist eine seltene, autosomal-rezessive, fortschreitende, neurodegenerative Form der Ataxie, die im Verlauf meist zum Verlust der Gehfähigkeit und letztendlich zum vorzeitigen Tod führt. Die Frühsymptome der Friedreich-Ataxie manifestieren sich in der Regel im Kindes- oder Jugendalter. Um eine rechtzeitige Diagnose, die Überweisung an spezialisierte Zentren und den Zugang zu potenziellen Therapien sicherzustellen, ist es wichtig, das Bewusstsein für die Friedreich-Ataxie sowie für ihre Symptome und Begleiterkrankungen zu schärfen. Diese CME richtet sich daher an alle Facharztgruppen mit dem Ziel, Awareness für die Friedreich-Ataxie als mögliche Ursache für diverse Symptomatiken und ihre Therapiemöglichkeiten zu schaffen.
Kursinhalt
Inhaltsverzeichnis
Vorwort
Die Friedreich-Ataxie ist eine seltene, autosomal-rezessive, fortschreitende, neurodegenerative Form der Ataxie, die im Verlauf meist zum Verlust der Gehfähigkeit und letztendlich zum vorzeitigen Tod führt. Benannt wurde die Friedreich-Ataxie nach dem deutschen Pathologen, Internisten und Neurologen Nicolaus Friedreich, der die Krankheit erstmals 1863 in Heidelberg dokumentierte. Die Frühsymptome der Friedreich-Ataxie, zu denen vor allem ein fortschreitender Verlust der Koordination (Gang-, Rumpf- und Zeigeataxie) gehört, manifestieren sich in der Regel im Kindes- oder Jugendalter. Die Friedreich-Ataxie geht mit erheblichen Auswirkungen auf die Lebensqualität der Patienten und ihrer Familien einher. Um eine rechtzeitige Diagnose, die Überweisung an spezialisierte Zentren und den Zugang zu potenziellen Therapien sicherzustellen, ist es wichtig, das Bewusstsein für die Friedreich-Ataxie sowie für ihre Symptome und Begleiterkrankungen wie die Kardiomyopathie und Skoliose zu schärfen [1]. Diese CME richtet sich daher an alle Facharztgruppen mit dem Ziel, Awareness für die Friedreich-Ataxie als mögliche Ursache für diverse Symptomatiken und ihre Therapiemöglichkeiten zu schaffen.
Hintergrund
Bei den hereditären Ataxien handelt es sich um eine klinisch und genetisch heterogene Gruppe von Erkrankungen, die phänotypisch durch Gang-, Rumpf- und Zeigeataxie sowie Dysarthrie und Okulomotorikstörungen gekennzeichnet sind. Häufig kommt es zu einer Atrophie des Kleinhirns. Hereditäre Ataxien können autosomal-dominant, autosomal-rezessiv, X-chromosomal oder mitochondrial vererbt werden. [2] Die häufigste autosomal-rezessive Ataxie ist die Friedreich-Ataxie, die meist im Alter zwischen 5 und 15 Jahren beginnt [3,4], wobei das Erkrankungsalter variieren kann und in manchen Fällen auch erst im Erwachsenenalter liegt. Ein Erkrankungsalter vor dem 25. Lebensjahr wird als typisch klassifiziert. Angaben zur Prävalenz der Friedreich-Ataxie in Deutschland variieren zwischen 1300 und 1800 Fällen [5].
Die Friedreich-Ataxie ist eine Multisystemerkrankung, die u.a. das Nerven-, Muskel-Skelett-, Herz- sowie das endokrine Pankreassystem betrifft [6]. Aufgrund der fortschreitenden und behindernden Symptome hat die Friedreich-Ataxie in der Regel einen erheblichen Einfluss auf die Lebensqualität [7]. Die meisten von der Friedreich-Ataxie betroffenen Menschen sind innerhalb von 10-20 Jahren nach Auftreten der ersten Symptome an den Rollstuhl gebunden [8]. Mit geeigneter und zielgerichteter Versorgung nach der Rollstuhlbindung können die Betroffenen aber auch noch viele Jahre leben [9]. Im Durchschnitt versterben Patienten mit Friedreich-Ataxie bereits mit 37 Jahren [9].
Genetische Ursache
Die Friedreich-Ataxie wird autosomal-rezessiv vererbt und ist in ca. 96% der Fälle durch eine homozygote GAA-Trinukleotid-Repeat-Expansion im ersten Intron des Frataxin-Gens (FXN) auf Chromosom 9 bedingt. In ca. 4% der Fälle liegt eine GAA-Trinukleotid-Repeat-Expansion auf einem Allel zusammen mit einer anderen inaktivierenden Mutation, z. B. eine Missense-, Nonsense-, Spleiß- oder Insertions-/Deletionsvariante, vor [10-12]. Die GAA-Trinukleotid-Repeat-Expansion reduziert die FXN-Expression und führt zu verringerten Spiegeln von Frataxin [11] (Abb. 1), einem hochkonservierten Protein, das mit der inneren Mitochondrienmembran in Verbindung steht [13]. Patienten mit Friedreich-Ataxie weisen unterschiedliche Ausmaße der verbliebenen Frataxin-Expression auf [13]. Die Länge der GAA-Wiederholung umfasst zwischen 66 und 1700 Tripletts und korreliert mit dem Erkrankungsalter [14] und dem Krankheitsverlauf [15-20].
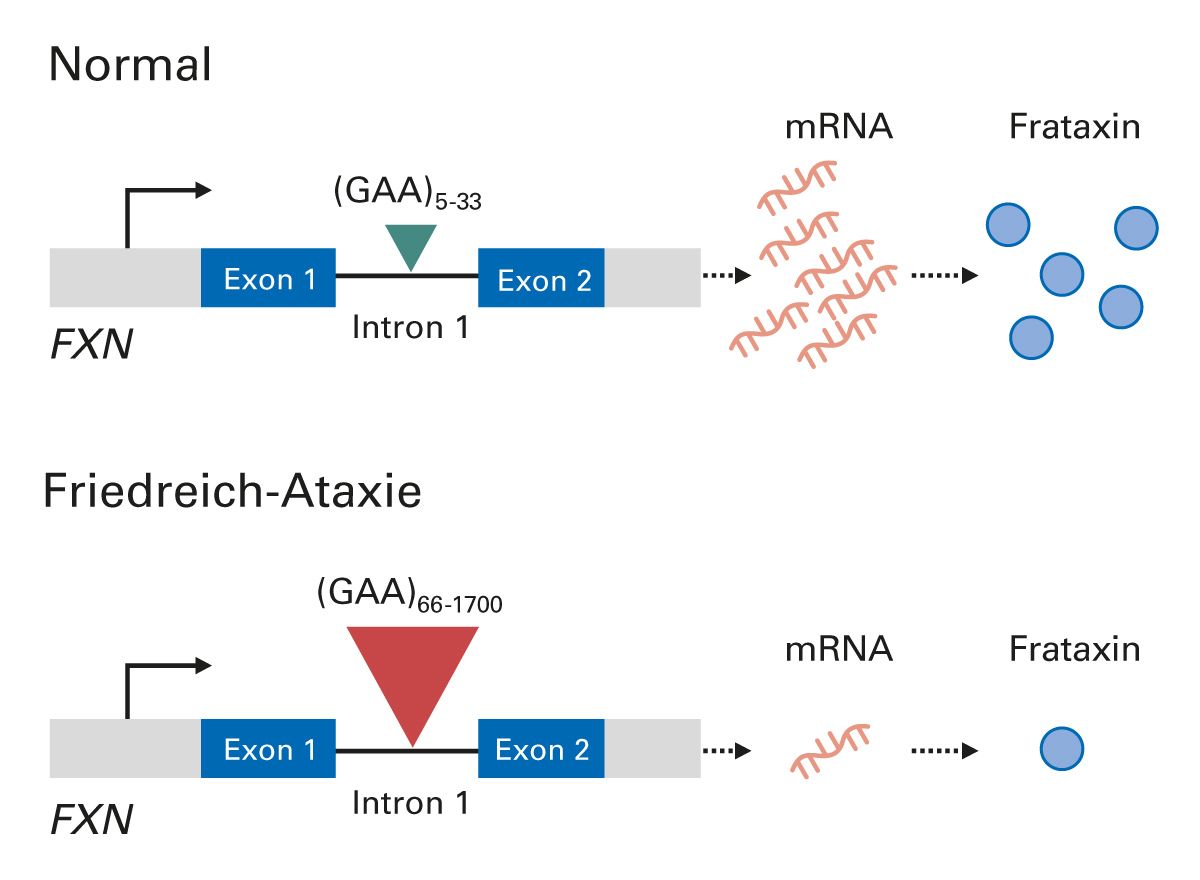
Abb. 1: Molekulare Ursache der Friedreich-Ataxie. Angepasst aus [20]
Pathologie
Das Frataxin-Protein spielt eine wesentliche Rolle im mitochondrialen Eisenstoffwechsel und der ATP-Produktion [22]. Ein Mangel führt zu einer hohen Eisenkonzentration in den Mitochondrien, was die Bildung toxisch wirkender freier reaktiver Sauerstoffspezies (ROS) fördert und mit vermehrtem oxidativem Stress einhergeht [23].
Die Gewebe mit der höchsten Frataxin-Expression sind die Hinterstränge, kortikospinalen und spinozerebellären Bahnen und der Nucleus dentatus im Kleinhirn im zentralen Nervensystem sowie die Spinalganglien und sensorischen peripheren Nerven im peripheren Nervensystem. Außerhalb des Nervensystems zeigte sich eine hohe Expression von Frataxin vor allem im Herzen, in den Beta-Zellen des Pankreas, in der Leber, der Skelettmuskulatur, im Thymus, der Haut, den Zähnen und im braunen Fettgewebe [13,24-27]. Somit führt ein Mangel an Frataxin zu mitochondrialer Dysfunktion und der Friedreich-Ataxie-Pathologie [28-30].
Diagnose
Schwierigkeiten beim Erkennen der Anzeichen von Friedreich-Ataxie können zu einer verzögerten Diagnose, Überweisung an spezialisierte Zentren und Zugang zu potenziellen Therapien führen [1]. Die Diagnose von Friedreich-Ataxie erfolgt typischerweise im Kindes- oder Jugendalter auf der Grundlage eines klinischen Verdachts bei Symptomen (Tab. 1) und wird durch einen spezifischen Gentest gesichert, der eine Mutation im FXN-Gen identifiziert [31]. Der Gentest kann als Teil der Regelversorgung der gesetzlichen Krankenversicherungen vom Arzt veranlasst werden. Eine spät einsetzende Friedreich-Ataxie kann aufgrund der manchmal atypischen klinischen Präsentation schwieriger zu erkennen sein [26]. Diagnostische Maßnahmen wie die Elektrophysiologie oder eine MR-Tomographie des Gehirns und Rückenmarks können die Diagnose stützen [32].
Klinisches Bild
Aus klinischer Sicht ist die Friedreich-Ataxie eine neurologische Erkrankung mit mehreren Multisystemmanifestationen, die fortschreiten und zu erheblichen Behinderungen führen können (Abb. 2) [34,35]. Die neurologischen Symptome der Friedreich-Ataxie sind bedingt durch eine Hinterstrangdegeneration, eine axonale Neuropathie und in geringerem Ausmaß eine zerebelläre Dysfunktion. Letztere manifestiert sich häufig erst später im Krankheitsverlauf. Klinisch-neurologisch zeigen sich typischerweise eine Gang-, Rumpf- und Zeigeataxie, Dysarthrie, Areflexie, Pallhypästhesie und, im Krankheitsverlauf, eine meist distal betonte Muskelatrophie, Kraftminderung der Extremitäten, Spastik und Pyramidenbahnzeichen sowie Dystonien [31,34]. Atypische Präsentationen mit erhaltenen Muskeleigenreflexen oder Spastizität mit minimaler oder keiner Ataxie können insbesondere bei Patienten mit spät einsetzender Friedreich-Ataxie nach dem 25. Lebensjahr auftreten [36]. Die zunehmende Verschlechterung trägt im fortgeschrittenen Stadium zu schweren Behinderungen bei [37]. Eine spät einsetzende Friedreich-Ataxie hat oft eine bessere Prognose und einen milderen Krankheitsverlauf [1,38].
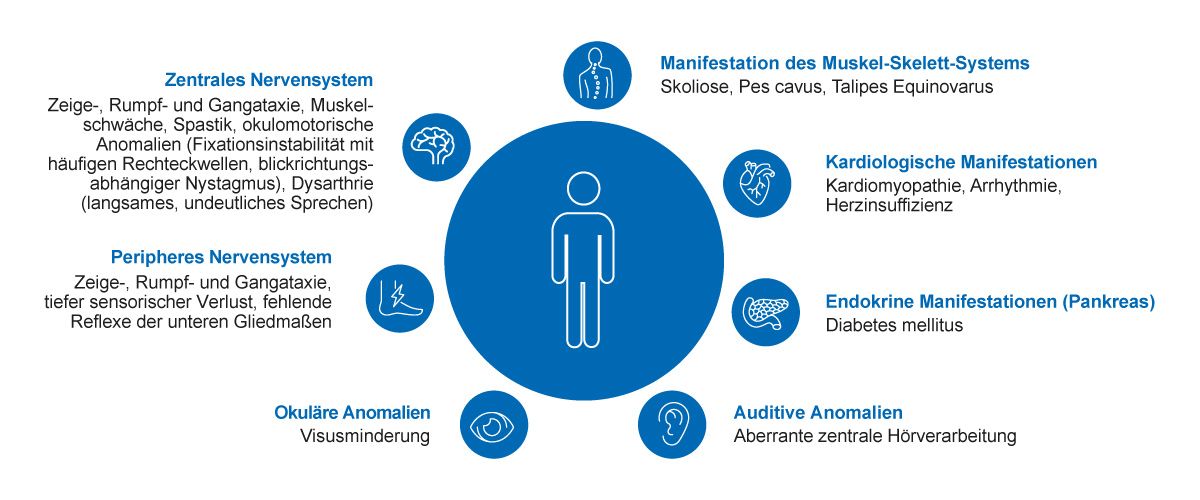
Abb. 2: Friedreich-Ataxie ist eine Multisystemerkrankung, die durch Läsionen sowohl im peripheren als auch im zentralen Nervensystem bedingt ist [34,35]
Friedreich-Ataxie und Kardiomyopathie
Kardiomyopathie tritt bei zwei Dritteln aller von Friedreich-Ataxie betroffenen Patienten auf. Sie ist komplikativ mit Herzinsuffizienz und Arrhythmien, meist Vorhofflimmern oder Vorhofflattern, assoziiert und ist mit 60 % die häufigste Todesursache bei Friedreich-Ataxie [39,35,40]. Kennzeichnend für die Friedreich-Ataxie-assoziierte Kardiomyopathie sind invertierte T-Wellen im EKG und eine linksventrikuläre und septale Hypertrophie in der Echokardiographie [41,42]. Die Ejektionsfraktion ist initial normal und nimmt erst später im Verlauf der Erkrankung ab. Ein Kardio-MRT ist für eine genauere und zuverlässigere Beurteilung der Septumhypertrophie und der linksventrikulären Gesamtmasse geeignet. Im Gegensatz zur Echokardiographie kann mithilfe des Kardio-MRTs auch schon eine kardiale Fibrose objektiviert werden [43]. Die Behandlung von Herzanomalien erfordert einen individuellen und interdisziplinären Ansatz gemeinsam mit kontinuierlicher kardiologischer Betreuung bei Patienten mit Friedreich-Ataxie, basierend auf dem Fortschreiten der Herz- und neurologischen Erkrankung, dem Alter sowie der Art der Arrhythmie oder der Herzinsuffizienz [9].
Friedreich-Ataxie und Diabetes
Etwa 10 % der Patienten mit Friedreich-Ataxie entwickeln einen Diabetes mellitus, in der Regel viele Jahre nach dem Auftreten der ersten Symptome, wobei die Schätzungen zur Prävalenz sehr unterschiedlich sind [44]. Diabetes kann einen unabhängigen negativen Einfluss auf das Wohlbefinden von Patienten mit Friedreich-Ataxie haben [18]. Die Patientenversorgung sollte daher standardmäßig alle 1-2 Jahre Diabetestests, z. B. den HbA1c umfassen [45]. Sobald Diabetes mellitus bei einem Patienten mit Friedreich-Ataxie bestätigt ist, müssen bei der individuellen Behandlung durch einen Endokrinologen patientenspezifische Parameter wie Alter, Komorbiditäten und Schweregrad des Diabetes mellitus berücksichtigt werden [31].
Friedreich-Ataxie und Skoliose
Studien haben gezeigt, dass nach klinischer Beurteilung bei etwa zwei Dritteln der Patienten mit Friedreich-Ataxie eine Skoliose vorliegt, nach radiologischer Beurteilung sogar bei bis zu 100 % [46,47]. Insbesondere während der Pubertät kommt es häufig zu einem raschen Fortschreiten der Skoliose [20]. Die Diagnose einer Skoliose wird durch die Bestimmung des Cobb-Winkels, welcher zur Messung einer Wirbelsäulenverkrümmung in der Frontal- oder Sagittalebene herangezogen wird, auf antero-posterioren und lateralen Röntgenaufnahmen der gesamten Wirbelsäule bestätigt [48]. Die Überwachung des Cobb-Winkels ist von wesentlicher Bedeutung und ist ein wesentlicher Faktor in der Planung, wann und welche Art von Intervention erforderlich ist [49]. Bracing ist eine nicht-chirurgische Option und kann zur Behandlung von Skoliosepatienten mit Krümmungen von 25-45 Grad erwogen werden [49,50]. Bei Krümmungen > 45 Grad und erheblichen Einschränkungen auf die Lebensqualität und Alltagskompetenz durch die Skoliose ist eine Operation meist indiziert [31]. Die präoperative Beurteilung sollte möglichst nach einer multidisziplinären Evaluation erfolgen. Hier sollte besonders die Kardiomyopathie beachtet werden und die Evaluation sollte kardiale Tests umfassen [50].
Management von Friedreich-Ataxie
Im Fall eines positiven bestätigenden Gentests kann ggf. eine genetische Beratung der Geschwister erwogen werden [9]. Zur vollständigen Beurteilung sollten Patienten mit einem positiven Gentest an lokale/regionale Friedreich-Ataxie-Spezialisten (Neurologen/Ataxiezentren) überwiesen werden [31]. Da Friedreich-Ataxie eine Multisystemerkrankung ist, erfordert die Betreuung eines Patienten mit Friedreich-Ataxie ein interdisziplinäres Behandlungsteam (Abb. 3).
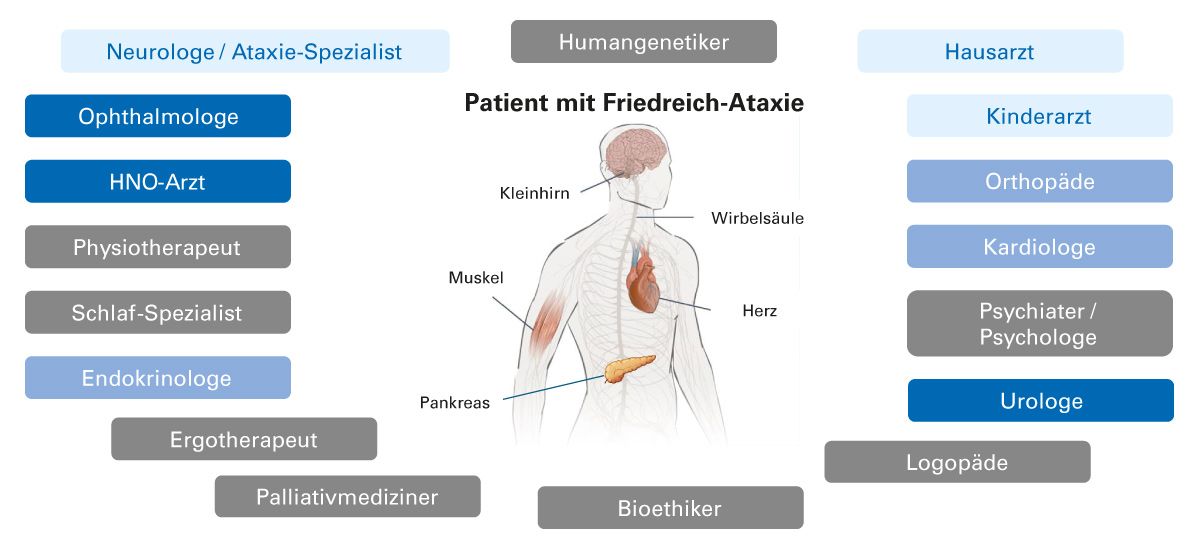
Abb. 3: In die Betreuung eines Patienten mit Friedreich-Ataxie sollte ein interdisziplinäres Behandlungsteam involviert sein [31]
Beurteilung des Schweregrades
Für die Beurteilung der neurologischen Dysfunktion und des klinischen Fortschreitens stehen verschiedene klinische Bewertungsskalen zur Verfügung. Die beiden meist genutzten Skalen sind die Scale for the Assessment and Rating of Ataxia (SARA) und die modifizierte Friedreich´s Ataxia Rating Scale (mFARS). Die SARA wurde nicht speziell für Friedreich-Ataxie entwickelt, in longitudinalen Beobachtungsstudien zeigte sie jedoch eine gute Eignung zur Erfassung der Progredienz der Ataxie bei Patienten mit Friedreich-Ataxie [4,51]. Die Skala besteht aus 8 Items zu Gangataxie, Standataxie, Rumpfataxie (Sitzen), Sprechen, Finger-Folge-Test, Finger-Nase-Versuch, Diadochokinese und Knie-Hacke-Versuch. Der Gesamtscore beträgt 40 Punkte [52]. Im Vergleich zur modifizierten Friedreich’s Ataxia Rating Scale (mFARS) wird hier die Gangtestung proportional höher bewertet, so dass die SARA für Patienten mit drohendem Verlust der Gehfähigkeit möglicherweise besser geeignet ist [53]. Der Zeitaufwand für SARA liegt zwischen 10 und 15 Minuten. Die mFARS wurde speziell für die Friedreich Ataxie entwickelt und ist eine klinische Skala über 4 Domänen (Bulbärmuskulatur, Koordination der oberen und unteren Extremitäten, Stabilität der aufrechten Körperhaltung), deren Gesamtscore 93 Punkte beträgt. Dabei korrelieren höhere Werte mit einer schlechteren neurologischen Funktion [54]. Ausgewählte Beispiele und mögliche klinische Auswirkungen sind in der Tabelle 2 dargestellt. In der mFARS wird die Standtestung proportional höher gewertet als in der SARA, so dass der mFARS für Patienten in sehr frühen Ataxiestadien möglicherweise besser geeignet ist [55,53]. SARA und mFARS werden als primärer Endpunkt in der Forschung und in klinischen Studien verwendet. Im klinischen Alltag ist die mFARS mit einem Zeitaufwand von 30 min jedoch weniger geeignet. Die psychometrischen Qualitäten der Skalen sind in vielerlei Hinsicht vergleichbar und die Wahl hängt z. T. von der zu untersuchenden Population ab [52-54].
Zur Beurteilung der Einschränkungen im Alltag ist insbesondere die Activities of Daily Living (ADL) Skala geeignet, welche im Rahmen eines strukturierten Interviews erhoben wird. Dieses Patient Reported Outcome Measure zeigte sogar eine höhere Sensitivität als die SARA für Progredienz der Erkrankung [51].
Therapieoptionen
Symptomorientierte Maßnahmen und die Behandlung von internistischen Begleiterkrankungen und Skelettdeformationen haben einen wichtigen Stellenwert in der Therapie der Friedreich-Ataxie. Die Therapiemöglichkeiten umfassen nichtmedikamentöse Verfahren wie Physio- und Ergotherapie, Logopädie, Bein- oder Fußstützen und Hilfsmittel wie Rollator und Rollstuhl sowie medikamentöse Therapeutika. Insbesondere die Physiotherapie hat auch einen verlaufsmodifzierenden Effekt [9,31,57,58]. In Beobachtungsstudien zeigte sich bei Patienten mit zerebellärer Ataxie nach einem intensiven 4-wöchigen Training eine signifikante Verbesserung der Ataxie, welche ungefähr einer natürlichen Krankheitsprogredienz von 2-4 Jahren entspricht [57,58].
Eine symptomatisch-orientierte Behandlung kann mit den nachfolgenden medikamentösen Optionen erfolgen: beispielsweise Baclofen, Tizanidin und Onabotulinumtoxin A zur Behandlung von Spastik, Gabapentin und Pregabalin bei neuropathischen Schmerzen, Anticholinergika wie Oxybutynin bei Harndrang sowie Beta-Blocker, ACE-Inhibitoren und Antiarrhythmika zur Therapie von Herz-Kreislauf-Erkrankungen [31].
Am 9. Februar 2024 wurde mit Omaveloxolon (Abb. 4) erstmals ein Medikament zur Behandlung der Friedreich-Ataxie bei Patienten ab 16 Jahren in Europa zugelassen [63]. Präklinische Daten deuten darauf hin, dass Omaveloxolon die Mitochondrienfunktion verbessern und oxidativen Stress reduzieren kann. An der zellulären Reaktion auf oxidativen Stress sind der Transkriptionsfaktor Nrf2 und das zytoplasmatische Protein Keap1 beteiligt. Unter normalen physiologischen Bedingungen ist Nrf2 an Keap1 gebunden und wird im Cytosol abgesondert. Bei oxidativem Stress führt die Oxidation von Keap1 zur Freisetzung und Translokation von Nrf2 in den Zellkern, wo es die Expression von Stressreaktionsgenen induziert (Abb. 5a). Bei Patienten mit Friedreich-Ataxie ist der Nrf2-Signalweg beeinträchtigt, so dass Nrf2 bei oxidativem Stress nicht in den Zellkern gelangen kann, um dort die notwendigen Stressreaktionsgene zu aktivieren (Abb. 5b) [28-30].
Abb. 4: Strukturformel von Omaveloxolon
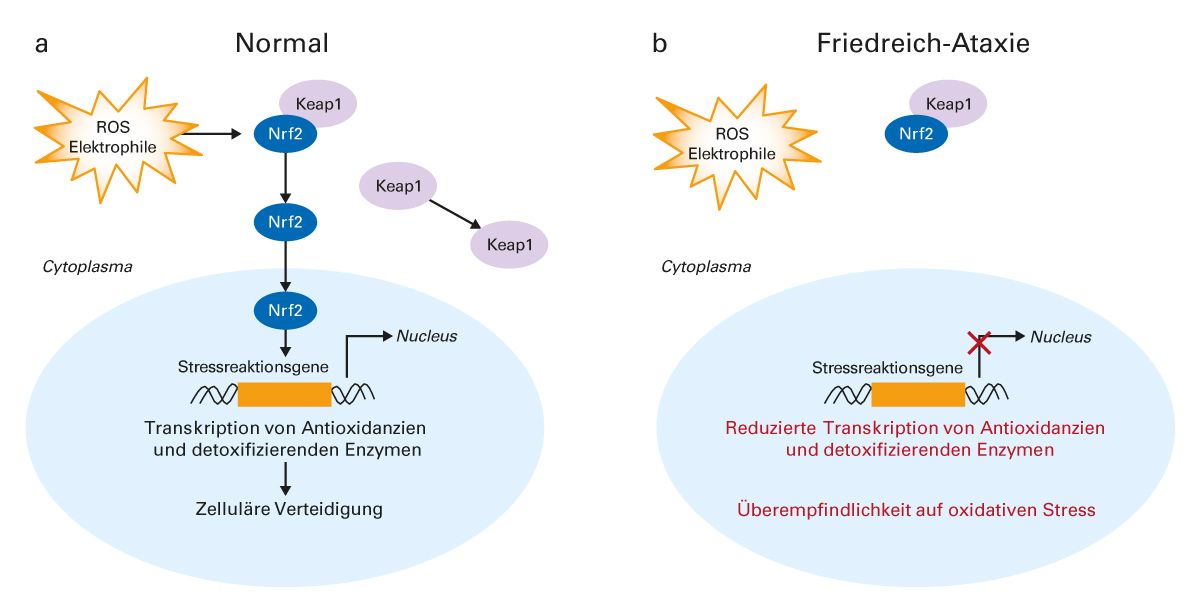
Abb. 5: Beeinträchtigung des Nrf2-Signalwegs bei Friedreich-Ataxie
Keap1, Kelch-like ECH-associated protein 1; Nrf2, Nuclear factor (Erythroid-derived 2) related factor 2; ROS, reaktive Sauerstoff-Spezies [28-30].
Omaveloxolon ist ein Nrf2-Aktivator, der den Keap1-vermittelten Abbau von Nrf2 stört, was in präklinischen Modellen zu Verbesserungen der mitochondrialen Dysfunktion, oxidativem Stress und Entzündungen führte (Abb. 6) [59-61]. Der genaue Wirkmechanismus ist noch unbekannt.
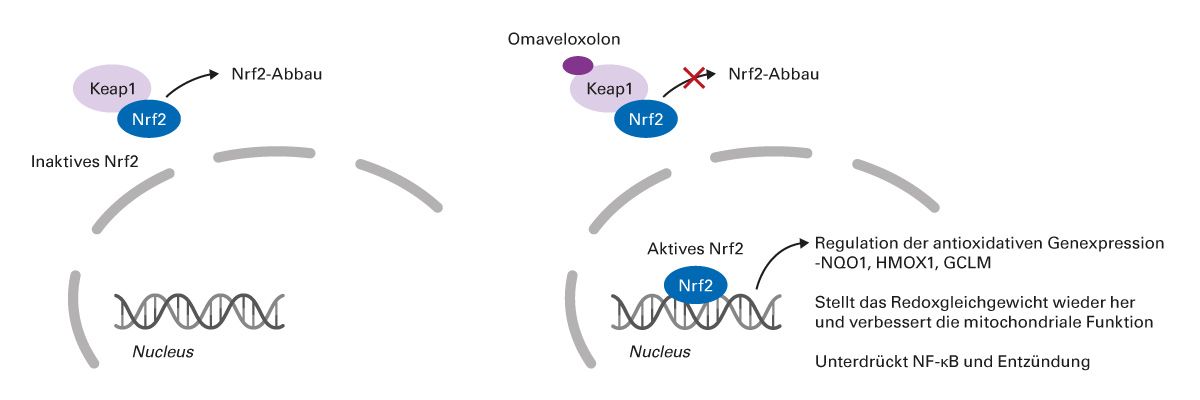
Abb. 6: Vermuteter Wirkmechanismus von Omaveloxolon auf Basis präklinischer Modelle [59-61].
GCLM, regulatorische Untereinheit der Glutamat-Cystein-Ligase; HMOX1, Hämoxygenase 1; KEAP1, Kelch-like ECH-associated protein 1; NF-κB, nuclear factor kappa-B; NQO1, NAD(P)H-Dehydrogenase [Chinon] 1; NRF2/Nrf2, nuclear factor erythroid 2–related factor 2
Omaveloxolon ist für die Behandlung der FA bei Patienten ab 16 Jahren zugelassen und die empfohlene Dosis beträgt 150 mg einmal täglich [63]. Die Zulassung erfolgte auf der Grundlage der Ergebnisse aus der klinischen Phase-II-Studie MOXIe, einer internationalen, doppelblinden, placebokontrollierten, randomisierten Parallelgruppenstudie. Eingeschlossen wurden 103 Patienten im Alter zwischen 16 und 40 Jahren mit genetisch bestätigter Friedreich-Ataxie, die auf der mFARS einen Wert zwischen 20 und 80 Punkten erreichten. Die Studienteilnehmer erhielten 1:1-randomisiert Omaveloxolon (150 mg einmal täglich) oder Placebo. Der primäre Endpunkt war die Differenz im mFARS-Wert zwischen Omaveloxolon und Placebo Gruppen in Woche 48 [62], wobei in den klinischen Studien zu Omaveloxolon die mFARS zusätzliche Fragen zur Bulbärmuskulatur beinhaltet und somit auf einen Gesamtscore von 99 Punkten kommt [62]. Die Behandlung mit Omaveloxolon verbesserte die neurologische Funktion signifikant, gemessen an der Reduktion der mFARS-Ausgangswerte im Vergleich zu Placebo nach 48 Behandlungswochen in der vollständigen Analysegruppe (FAS) (Δ -2,4 Punkte; p=0,014) (Abb. 7) [62]. Das Ausmaß der beobachteten Verbesserungen entspricht etwa der Verzögerung der Krankheitsprogression um 1-2 Jahre [56].
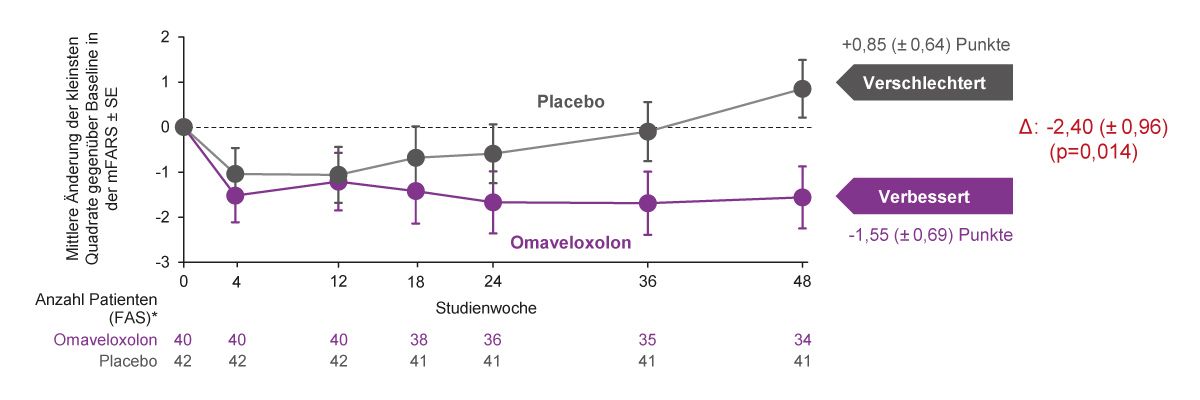
Abb. 7: Primärer Endpunkt der MOXIe-Studie: Änderung vom Ausgangswert im mFARS nach 48 Wochen [62].(*Patienten mit Pes cavus wurden aus dem Full-Analysis-Set ausgeschlossen (FAS; n=82); Δ: Unterschied zwischen
den Behandlungsgruppen in Woche 48 (Omaveloxolon – Placebo); FAS, Full Analysis Set; SE, Standardfehler)
Sensitivitätsanalysen, bei denen auch Patienten mit Pes cavus berücksichtigt wurden, bestätigten die Analyse des primären Endpunkts. Die Verbesserungen unter Omaveloxolon im mFARS waren in allen Subgruppen (Stratifizierung nach Alter, Geschlecht, GAA-Repeatlänge, Status der Gehfähigkeit) konsistent. Omaveloxolon wurde im Allgemeinen gut vertragen, es gab nur wenige Therapieabbrüche oder schwerwiegende unerwünschte Ereignisse [62]. Allerdings war eine sehr häufige Nebenwirkung der Anstieg der Aminotransferasen, der sich meist nach ca. 12 Wochen regredient zeigte. Ab einem Anstieg über das 5-Fache des oberen Referenzwertes, oder einem Anstieg auf das über 3-Fache des oberen Referenzwerts bei gleichzeitigem Anstieg auf das über 2-Fache des Bilirubins sollte daher die Therapie mit Omaveloxolon pausiert werden, bis sich die Leberwerte normalisiert haben. Ebenfalls zeigte sich unter Therapie mit Omaveloxolon häufig ein Anstieg der Cholesterinwerte (Gesamtcholesterin und LDL). Weniger häufig kam es auch zu einem Anstieg des natriuretischen Peptids Typ B (BNP), meist ohne weitere Anzeichen einer Herzinsuffizienz. Aufgrund dessen sollte eine Kontrolle der Leberwerte, Lipidwerte und des BNP-Werts vor Beginn der Behandlung durchgeführt werden. Es wird ein regelmäßiges Monitoring der Leberwerte empfohlen: in den ersten 3 Monaten wird bei stabilen Werten mindestens eine monatliche Kontrolle empfohlen, danach mindestens alle 3 Monate. Weitere Nebenwirkungen beinhalten Kopfschmerzen, Gewichtsabnahme und verminderter Appetit, Übelkeit und Erbrechen, Diarrhoe, Rückenschmerzen [63]. Omaveloxolon ist ein Substrat von CYP3A4. Daher kann die gleichzeitige Anwendung von starken oder moderaten CYP3A4-Inhibitoren oder CYP3A4-Induktoren die Pharmakokinetik von Omaveloxolon beeinflussen und sollte nach Möglichkeit vermieden werden [63].
Die Ergebnisse einer offenen Verlängerungsstudie von MOXIe wiesen darauf hin, dass die motorischen und bulbären Funktionen unter Omaveloxolon längerfristig verbessert werden können und ein früher Einsatz möglicherweise dabei von Vorteil ist. An der zweiarmigen Verlängerungsstudie nahmen 34 Patienten aus dem Omaveloxolon-Arm und 39 Patienten aus dem Placebo-Arm von MOXIe teil. Alle erhielten ab Woche 52 Omaveloxolon 150 mg einmal täglich über einen Zeitraum bis Woche 144-168. Die Verbesserungen im mFARS blieben in der Gruppe, die bereits bis Woche 48 Omaveloxolon erhalten hatte, gegenüber dem Vergleichsarm über diesen längeren Zeitraum bestehen (Δ −2,91 ± 1,44 Punkte) [64]. Die Verlängerungsstudie wurde bis Juni 2024 fortgesetzt.
Das klinische Studienprogramm für Omaveloxolon wird durch Post-hoc-Analysen mit Propensity-Score-Matching und Analysen mit verzögertem Therapiebeginn unterstützt. In der Analyse mit Propensity-Score-Matching wurde über einen Zeitraum von drei Jahren ein Nutzen bei den mit Omaveloxolon behandelten Patienten im Vergleich zu einer unbehandelten Kohorte mit natürlichem Verlauf festgestellt [65]. Diese exploratorische Analyse sollte jedoch mit Vorsicht interpretiert werden, da die außerhalb einer kontrollierten Studie erhobenen Daten nur begrenzt aussagekräftig sind und es zu Störeinflüssen kommen kann.
Weitere Wirkstoffe zur Therapie der FA befinden sich aktuell in der klinischen Entwicklung. Neben Substanzen, die wie Omaveloxolon die Mitochondrienfunktion verbessern und oxidativen Stress reduzieren sollen, haben andere Kandidaten die Modulation der Frataxin-kontrollierten Signalwege, den Ersatz von Frataxin, die Erhöhung der FXN-Genexpression oder das Ersetzen des FXN-Gens zum Ziel [66].
Fazit
Friedreich-Ataxie ist eine seltene hereditäre, fortschreitende, neurodegenerative Krankheit mit erheblichen Auswirkungen auf die Lebensqualität der Patienten und ihrer Familien [3,4]. Die ersten Symptome treten meistens im Alter zwischen 5 und 15 Jahren auf, jedoch weisen sowohl das Erkrankungsalter als auch der Krankheitsverlauf eine erhebliche inter-individuelle Variabilität auf [3,4]. Ursache ist in den meisten Fällen eine homozygote GAA-Trinukleotid-Repeat-Expansion im FXN-Gen, was zu einem Mangel des Proteins Frataxin und somit zu einer mitochondrialen Dysfunktion führt [10-14]. Die Diagnose erfolgt auf der Grundlage eines klinischen Verdachts bei entsprechender Symptomatik und wird molekulargenetisch durch die Detektion einer Mutation im FXN-Gen gesichert [31]. Die modifizierte Friedreich’s Ataxia Rating Scale (mFARS) und die Scale for Assessment and Rating of Ataxia (SARA) werden zur Einordnung des Schweregrads der Ataxie verwendet, wobei geringere Werte bessere Funktionen bedeuten [4,51]. Die Behandlung der Friedreich-Ataxie erfordert ein interdisziplinäres Behandlungsteam. Seit kurzem ist Omaveloxolon als erste medikamentöse Therapie für die Behandlung der Friedreich-Ataxie bei Patienten ab 16 Jahren in der EU zugelassen [63]. Dabei handelt es sich um einen Nrf2-Aktivator, der den Keap1-vermittelten Abbau von Nrf2 stört, was in präklinischen Modellen zu Verbesserungen der mitochondrialen Dysfunktion und verringertem oxidativem Stress führte [59-61]. Analysen der klinischen Zulassungsstudie haben eine Verlangsamung der Behinderungsprogression unter Omaveloxolon im Vergleich zum natürlichen Verlauf bei insgesamt guter Verträglichkeit gezeigt [62-65]. Symptomorientierte Begleittherapien bleiben weiterhin ein wichtiges Standbein in der Patientenversorgung.
Literatur
- Indelicato E, Nachbauer W, Eigentler A et al. (2020) Onset features and time to diagnosis in Friedreich’s Ataxia. Orphanet J Rare Dis 15 (1):198. doi:10.1186/s13023-020-01475-9
- Jayadev S, Bird D Thomas et al. (2013) Hereditary ataxias: overview. Genet Med 2013 Sep;15(9):673-83. doi: 10.1038/gim.2013.28. Epub 2013 Mar 28.
- Reetz K, Dogan I, Costa AS et al. (2015) Biological and clinical characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS) cohort: a cross-sectional analysis of baseline data. Lancet Neurol 14 (2):174-182. doi:10.1016/s1474-4422(14)70321-7
- Reetz K, Dogan I, Hilgers RD et al. (2016) Progression characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS): a 2 year cohort study. Lancet Neurol 15 (13):1346-1354. doi:10.1016/s1474-4422(16)30287-3
- Vankan P (2013) Prevalence gradients of Friedreich’s ataxia and R1b haplotype in Europe co-localize, suggesting a common Palaeolithic origin in the Franco-Cantabrian ice age refuge. J Neurochem 126 Suppl 1:11-20. doi:10.1111/jnc.12215
- Cook A, Giunti P (2017) Friedreich’s ataxia: clinical features, pathogenesis and management. Br Med Bull 124 (1):19-30. doi:10.1093/bmb/ldx034
- Giunti P, Greenfield J, Stevenson AJ et al. (2013) Impact of Friedreich’s Ataxia on health-care resource utilization in the United Kingdom and Germany. Orphanet J Rare Dis 8:38. doi:10.1186/1750-1172-8-38
- Fogel BL, Perlman S (2007) Clinical features and molecular genetics of autosomal recessive cerebellar ataxias. Lancet Neurol 6 (3):245-257. doi:10.1016/s1474-4422(07)70054-6
- Corben LA, Collins V, Milne S et al. (2022) Clinical management guidelines for Friedreich ataxia: best practice in rare diseases. Orphanet J Rare Dis 17 (1):415. doi:10.1186/s13023-022-02568-3
- Bürk K (2017) Friedreich Ataxia: current status and future prospects. Cerebellum Ataxias 4:4. doi:10.1186/s40673-017-0062-x
- Campuzano V, Montermini L, Moltò MD et al. (1996) Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271 (5254):1423-1427. doi:10.1126/science.271.5254.1423
- Galea CA, Huq A, Lockhart PJ et al. (2016) Compound heterozygous FXN mutations and clinical outcome in friedreich ataxia. Ann Neurol 79 (3):485-495. doi:10.1002/ana.24595
- Campuzano V, Montermini L, Lutz Y et al. (1997) Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum Mol Genet 6 (11):1771-1780. doi:10.1093/hmg/6.11.1771
- Dürr A, Cossee M, Agid Y et al. (1996) Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N Engl J Med 335 (16):1169-1175. doi:10.1056/nejm199610173351601
- Hanson E, Sheldon M, Pacheco B, Alkubeysi M, Raizada V (2019) Heart disease in Friedreich’s ataxia. World J Cardiol 11 (1):1-12. doi:10.4330/wjc.v11.i1.1
- La Pean A, Jeffries N, Grow C, Ravina B, Di Prospero NA (2008) Predictors of progression in patients with Friedreich ataxia. Mov Disord 23 (14):2026-2032. doi:10.1002/mds.22248
- Mateo I, Llorca J, Volpini V et al. (2004) Expanded GAA repeats and clinical variation in Friedreich’s ataxia. Acta Neurol Scand 109 (1):75-78. doi:10.1034/j.1600-0404.2003.00190.x
- McCormick A, Farmer J, Perlman S et al. (2017) Impact of diabetes in the Friedreich ataxia clinical outcome measures study. Ann Clin Transl Neurol 4 (9):622-631. doi:10.1002/acn3.439
- Peverill RE, Romanelli G, Donelan L et al. (2019) Left ventricular structural and functional changes in Friedreich ataxia – Relationship with body size, sex, age and genetic severity. PLoS One 14 (11):e0225147. doi:10.1371/journal.pone.0225147
- Rummey C, Flynn JM, Corben LA et al. (2021) Scoliosis in Friedreich’s ataxia: longitudinal characterization in a large heterogeneous cohort. Ann Clin Transl Neurol 8 (6):1239-1250. doi:10.1002/acn3.51352
- Prescribing Information FDA. Stand Februar 2023
- Delatycki MB, Bidichandani SI (2019) Friedreich ataxia- pathogenesis and implications for therapies. Neurobiol Dis 132:104606. doi:10.1016/j.nbd.2019.104606
- Chiang S, Kalinowski DS, Jansson PJ, Richardson DR, Huang ML (2018) Mitochondrial dysfunction in the neuro-degenerative and cardio-degenerative disease, Friedreich’s ataxia. Neurochem Int 117:35-48. doi:10.1016/j.neuint.2017.08.002
- González-Cabo P, Palau F (2013) Mitochondrial pathophysiology in Friedreich’s ataxia. J Neurochem 126 Suppl 1:53-64. doi:10.1111/jnc.12303
- Jiralerspong S, Liu Y, Montermini L, Stifani S, Pandolfo M (1997) Frataxin shows developmentally regulated tissue-specific expression in the mouse embryo. Neurobiol Dis 4 (2):103-113. doi:10.1006/nbdi.1997.0139
- Koeppen AH (2011) Friedreich’s ataxia: pathology, pathogenesis, and molecular genetics. J Neurol Sci 303 (1-2):1-12. doi:10.1016/j.jns.2011.01.010
- Koutnikova H, Campuzano V, Foury F et al. (1997) Studies of human, mouse and yeast homologues indicate a mitochondrial function for frataxin. Nat Genet 16 (4):345-351. doi:10.1038/ng0897-345
- Anzovino A, Chiang S, Brown BE et al. (2017) Molecular Alterations in a Mouse Cardiac Model of Friedreich Ataxia: An Impaired Nrf2 Response Mediated via Upregulation of Keap1 and Activation of the Gsk3β Axis. Am J Pathol 187 (12):2858-2875. doi:10.1016/j.ajpath.2017.08.021
- Petrillo S, Piermarini E, Pastore A et al. (2017) Nrf2-Inducers Counteract Neurodegeneration in Frataxin-Silenced Motor Neurons: Disclosing New Therapeutic Targets for Friedreich’s Ataxia. Int J Mol Sci 18 (10). doi:10.3390/ijms18102173
- Shan Y, Schoenfeld RA, Hayashi G et al. (2013) Frataxin deficiency leads to defects in expression of antioxidants and Nrf2 expression in dorsal root ganglia of the Friedreich’s ataxia YG8R mouse model. Antioxid Redox Signal 19 (13):1481-1493. doi:10.1089/ars.2012.4537
- Lynch DR, Schadt K, Kichula E, McCormack S, Lin KY (2021) Friedreich Ataxia: Multidisciplinary Clinical Care. J Multidiscip Healthc 14:1645-1658. doi:10.2147/jmdh.S292945
- Klockgether T. et al., Ataxien des Erwachsenenalters, S1-Leitlinine, 2023, in: Deutsche Gesellschaft für Neurologie (Hrsg.), Leitlinien für Diagnostik und Therapie in der Neurologie. Online: www.dgn.org/leitlininen (abgerufen am 03.05.2024).
- Reetz K, Dogan I, Hohenfeld C et al. (2018) Nonataxia symptoms in Friedreich Ataxia: Report from the Registry of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS). Neurology 91 (10):e917-e930. doi:10.1212/wnl.0000000000006121
- Parkinson MH, Boesch S, Nachbauer W, Mariotti C, Giunti P (2013) Clinical features of Friedreich’s ataxia: classical and atypical phenotypes. J Neurochem 126 Suppl 1:103-117. doi:10.1111/jnc.12317
- Williams CT, De Jesus O. Friedreich Ataxia. 2023 Aug 23. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2024.
- Bhidayasiri R, Perlman SL, Pulst SM, Geschwind DH (2005) Late-onset Friedreich ataxia: phenotypic analysis, magnetic resonance imaging findings, and review of the literature. Arch Neurol 62 (12):1865-1869. doi:10.1001/archneur.62.12.1865
- Fichera M, Castaldo A, Mongelli A et al. (2022) Comorbidities in Friedreich ataxia: incidence and manifestations from early to advanced disease stages. Neurol Sci 43 (12):6831-6838. doi:10.1007/s10072-022-06360-w
- Lecocq C, Charles P, Azulay JP et al. (2016) Delayed-onset Friedreich’s ataxia revisited. Mov Disord 31 (1):62-69. doi:10.1002/mds.26382
- Jensen MK, Bundgaard H (2012) Cardiomyopathy in Friedreich ataxia: exemplifying the challenges faced by cardiologists in the management of rare diseases. Circulation 125 (13):1591-1593. doi:10.1161/circulationaha.112.095364
- Tsou AY, Paulsen EK, Lagedrost SJ et al. (2011) Mortality in Friedreich ataxia. J Neurol Sci 307 (1-2):46-49. doi:10.1016/j.jns.2011.05.023
- Rossi M, Wainsztein N, Merello M (2021) Cardiac Involvement in Movement Disorders. Mov Disord Clin Pract 8 (5):651-668. doi:10.1002/mdc3.13188
- Weidemann F, Störk S, Liu D et al. (2013) Cardiomyopathy of Friedreich ataxia. J Neurochem 126 Suppl 1:88-93. doi:10.1111/jnc.12217
- Lynch DR, Regner SR, Schadt KA et al. (2012) Management and therapy for cardiomyopathy in Friedreich’s ataxia. Expert Rev Cardiovasc Ther 10 (6):767-777. doi:10.1586/erc.12.57
- Gucev Z, Tasic V, Jancevska A et al. (2009) Friedreich ataxia (FA) associated with diabetes mellitus type 1 and hyperthrophic cardiomyopathy. Bosn J Basic Med Sci 9 (2):107-110. doi:10.17305/bjbms.2009.2828
- Association AD (2021) Classification and Diagnosis of Diabetes: Standards of Medical Care in Diabetes-2021. Diabetes Care 44 (Suppl 1):S15-s33. doi:10.2337/dc21-S002
- Labelle H, Tohmé S, Duhaime M, Allard P (1986) Natural history of scoliosis in Friedreich’s ataxia. J Bone Joint Surg Am 68 (4):564-572
- Milbrandt TA, Kunes JR, Karol LA (2008) Friedreich’s ataxia and scoliosis: the experience at two institutions. J Pediatr Orthop 28 (2):234-238. doi:10.1097/BPO.0b013e318164fa79
- Sheehan DD, Grayhack J (2017) Pediatric Scoliosis and Kyphosis: An Overview of Diagnosis, Management, and Surgical Treatment. Pediatr Ann 46 (12):e472-e480. doi:10.3928/19382359-20171113-01
- El-Hawary R, Chukwunyerenwa C (2014) Update on evaluation and treatment of scoliosis. Pediatr Clin North Am 61 (6):1223-1241. doi:10.1016/j.pcl.2014.08.007
- Tsirikos AI, Smith G (2012) Scoliosis in patients with Friedreich’s ataxia. J Bone Joint Surg Br 94 (5):684-689. doi:10.1302/0301-620x.94b5.28391
- Reetz K, Dogan I, Hilgers RD et al. (2021) Progression characteristics of the European Friedreich’s Ataxia Consortium for Translational Studies (EFACTS): a 4-year cohort study. Lancet Neurol 20 (5):362-372. doi:10.1016/s1474-4422(21)00027-2
- Schmitz-Hübsch T, du Montcel ST, Baliko L et al. (2006) Scale for the assessment and rating of ataxia: development of a new clinical scale. Neurology 66 (11):1717-1720. doi:10.1212/01.wnl.0000219042.60538.92
- Rummey C, Harding IH, Delatycki MB et al. (2022) Harmonizing results of ataxia rating scales: mFARS, SARA, and ICARS. Ann Clin Transl Neurol 9 (12):2041-2046. doi:10.1002/acn3.51686
- Rummey C, Corben LA, Delatycki MB et al. (2019) Psychometric properties of the Friedreich Ataxia Rating Scale. Neurol Genet 5 (6):371. doi:10.1212/nxg.0000000000000371
- Subramony SH, May W, Lynch D et al. (2005) Measuring Friedreich ataxia: Interrater reliability of a neurologic rating scale. Neurology 64 (7):1261-1262. doi:10.1212/01.Wnl.0000156802.15466.79
- Patel M, Isaacs CJ, Seyer L et al. (2016) Progression of Friedreich ataxia: quantitative characterization over 5 years. Ann Clin Transl Neurol 3 (9):684-694. doi:10.1002/acn3.332
- Ilg W, Brötz D, Burkard S et al. (2010) Long-term effects of coordinative training in degenerative cerebellar disease. Mov Disord 25 (13):2239-2246. doi:10.1002/mds.23222
- Ilg W, Synofzik M, Brötz D et al. (2009) Intensive coordinative training improves motor performance in degenerative cerebellar disease. Neurology 73 (22):1823-1830. doi:10.1212/WNL.0b013e3181c33adf
- Abeti R, Baccaro A, Esteras N, Giunti P (2018) Novel Nrf2-Inducer Prevents Mitochondrial Defects and Oxidative Stress in Friedreich’s Ataxia Models. Front Cell Neurosci 12:188. doi:10.3389/fncel.2018.00188
- Cuadrado A, Rojo AI, Wells G et al. (2019) Therapeutic targeting of the NRF2 and KEAP1 partnership in chronic diseases. Nat Rev Drug Discov 18 (4):295-317. doi:10.1038/s41573-018-0008-x
- Probst BL, Trevino I, McCauley L et al. (2015) RTA 408, A Novel Synthetic Triterpenoid with Broad Anticancer and Anti-Inflammatory Activity. PLoS One 10 (4):e0122942. doi:10.1371/journal.pone.0122942
- Lynch DR, Chin MP, Delatycki MB et al. (2021) Safety and Efficacy of Omaveloxolone in Friedreich Ataxia (MOXIe Study). Ann Neurol 89 (2):212-225. doi:10.1002/ana.25934
- Fachinformation Omaveloxolon. Stand August 2024.
- Lynch DR, Chin MP, Boesch S et al. (2023) Efficacy of Omaveloxolone in Friedreich’s Ataxia: Delayed-Start Analysis of the MOXIe Extension. Mov Disord 38 (2):313-320. doi:10.1002/mds.29286
- Lynch DR, Goldsberry A, Rummey C et al. (2024) Propensity matched comparison of omaveloxolone treatment to Friedreich ataxia natural history data. Ann Clin Transl Neurol 11 (1):4-16. doi:10.1002/acn3.51897
- Friedreich’s Ataxis Research Alliance. Research Pipeline. https://www.curefa.org/research/research-pipeline. Abgerufen am: 21.5.2024
Tutorielle Unterstützung
Die tutorielle Unterstützung der Fortbildungsteilnehmer erfolgt durch unseren ärztlichen Leiter Dr. med. Alexander Voigt in Zusammenarbeit mit der arztCME-Redaktion. Inhaltliche Fragen können über das Kommentarfeld, direkt per Mail an service@arztcme.de oder via Telefon unter Tel.: +49(0)180-3000759 gestellt werden. Inhaltliche Fragen werden von unserem ärztlichen Leiter bzw. nach Rücksprache mit diesem und evtl. dem Autor auch von der arztCME-Redaktion beantwortet.
Technischer Support
Der technische Support der arztCME-Online-Akademie erfolgt durch geschulte Mitarbeiterinnen und Mitarbeiter des Betreibers health&media GmbH unter der E-Mail-Adresse technik@arztcme.de oder via Telefon unter Tel.: 49(0)180-3000759.